Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Integrantes do grupo: Ademir, André, Carine,Jéssica, Joice e Vanderlei
FIBROSE CÍSTICA Integrantes do grupo: Ademir, André, Carine,Jéssica, Joice e Vanderlei Professor: Leandro Tibiriçá Burgos Disciplina: Prescrição e controle do exercício para populações Especiais

2
INTRODUÇÃO: Neste trabalho abordaremos:FIBROSE CÍSTICA O que é;
Causas; Como detectar; Sintomas; Tratamento; Relação com atividade física; Apresentação de estudo de caso.

3
O QUE É FIBROSE CÍSTICA? A fibrose cística é uma doença genética, de caráter autossômico recessivo, que compromete o funcionamento de todos os órgãos e sistemas do organismo, através da alteração da função das glândulas exócrinas. É uma doença evolutiva que não tem cura. (ROCHA, MOREIRA & OLIVEIRA, 2004). É uma doença hereditária que causa o acúmulo de muco denso e pegajoso nos pulmões, no trato digestivo e outras áreas do corpo. É uma das doenças pulmonares crônicas mais comuns em crianças e adultos jovens. Essa é uma doença que envolve risco de vida. (
. É uma doença hereditária que causa o acúmulo de muco denso e pegajoso nos pulmões, no trato digestivo e outras áreas do corpo. É uma das doenças pulmonares crônicas mais comuns em crianças e adultos jovens. Essa é uma doença que envolve risco de vida. (")
4
A FC é mais comum na raça branca, que incide em uma para duas mil crianças nascidas vivas, enquanto na raça negra é de uma para quinze mil, e pouco frequente nos índios e orientais (PINHEIRO, 1998, apud KRÄMER, 2008).
.")
5
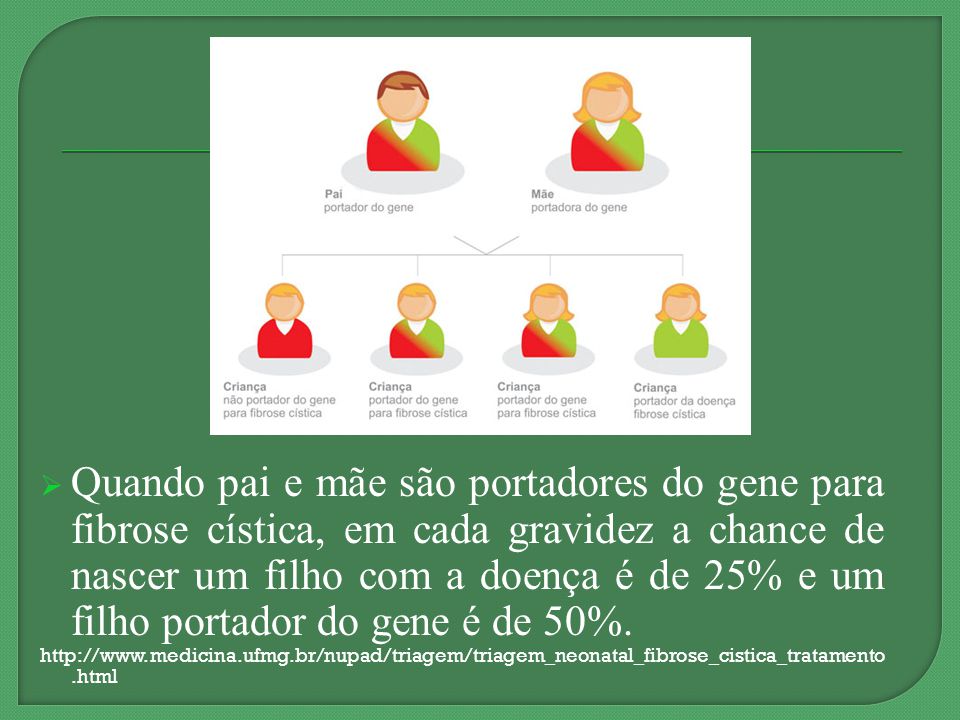
Quando pai e mãe são portadores do gene para fibrose cística, em cada gravidez a chance de nascer um filho com a doença é de 25% e um filho portador do gene é de 50%.
6
Se ambos os membros de um casal forem portadores do mesmo gene afetado, podem transmitir aos seus filhos um gene normal ou um gene alterado. Isto acontece ao acaso (de forma aleatória). Deste modo, cada filho ou filha de pais que são ambos portadores do mesmo gene alterado tem uma probabilidade de 25% (1 em cada 4) de herdar duas cópias alteradas do mesmo gene, uma de cada um dos pais, e assim vir a ser afetada pela doença. Isto também significa que há uma probabilidade de 75% (3 em cada 4) que a criança não seja afetada pela doença. Esta probabilidade mantém-se para cada gravidez e é igual para rapazes e raparigas. Há também uma probabilidade de 50% (2 em cada 4) de a criança herdar apenas uma cópia do gene alterado de um dos pais. Se isto acontecer a criança será um portador saudável tal como os pais. Finalmente, existe ainda uma probabilidade de 25% (1 em cada 4) de a criança herdar ambas as cópias normais de cada um dos pais. Neste caso o/a filho/a não terá a doença, nem será portador/a da mesma. Todas estas hipóteses ocorrem completamente ao acaso. As probabilidades mantêm-se as mesmas para cada gravidez e são as mesmas para rapazes e raparigas.
 de herdar duas cópias alteradas do mesmo gene, uma de cada um dos pais, e assim vir a ser afetada pela doença. Isto também significa que há uma probabilidade de 75% (3 em cada 4) que a criança não seja afetada pela doença. Esta probabilidade mantém-se para cada gravidez e é igual para rapazes e raparigas. Há também uma probabilidade de 50% (2 em cada 4) de a criança herdar apenas uma cópia do gene alterado de um dos pais. Se isto acontecer a criança será um portador saudável tal como os pais. Finalmente, existe ainda uma probabilidade de 25% (1 em cada 4) de a criança herdar ambas as cópias normais de cada um dos pais. Neste caso o/a filho/a não terá a doença, nem será portador/a da mesma. Todas estas hipóteses ocorrem completamente ao acaso. As probabilidades mantêm-se as mesmas para cada gravidez e são as mesmas para rapazes e raparigas.")
7
CAUSAS: A fibrose cística (FC) é causada por um gene defeituoso que faz com que o corpo produza um líquido anormalmente denso e pegajoso chamado muco. O muco se acumula nas passagens respiratórias dos pulmões e no pâncreas, órgão que ajuda a decompor e absorver o alimento. Essa coleta de muco pegajoso resulta em infecções pulmonares que colocam a vida em risco, além de problemas digestivos graves. A doença também pode afetar as glândulas sudoríparas e o sistema reprodutivo masculino.
 é causada por um gene defeituoso que faz com que o corpo produza um líquido anormalmente denso e pegajoso chamado muco. O muco se acumula nas passagens respiratórias dos pulmões e no pâncreas, órgão que ajuda a decompor e absorver o alimento. Essa coleta de muco pegajoso resulta em infecções pulmonares que colocam a vida em risco, além de problemas digestivos graves. A doença também pode afetar as glândulas sudoríparas e o sistema reprodutivo masculino.")
8
COMO DETECTAR: Seu diagnóstico é baseado nas manifestações clínicas associadas ao teste do suor alterado. Um alto nível de sal no suor do paciente é sinal da doença. O teste de DNA pode ou não identificar a mutação da fibrose cística. O teste procura por variações em um gene conhecido que causa a doença. Um teste de sangue está disponível para ajudar a detectar a FC. Outros testes usados para diagnosticar a FC incluem: O teste de tripsinogênio imunorreativo (TIR) é um teste padrão de triagem em recém nascidos para FC. Um alto nível de TIR sugere possível FC e requer mais testes.
 é um teste padrão de triagem em recém nascidos para FC. Um alto nível de TIR sugere possível FC e requer mais testes.")
9
COMO DETECTAR: Outros testes que identificam problemas que podem estar relacionados à fibrose cística incluem: Raio X ou tomografia computadorizada do tórax Teste de gordura nas fezes Testes de funcionamento dos pulmões Medição da função pancreática Teste de estimulação da secretina Tripsina e quimotripsina nas fezes Série do trato gastrointestinal superior (GI) e do intestino delgado
 e do intestino delgado.")
10
COMO DETECTAR: A maioria das crianças com FC é diagnosticada até 2 anos de idade. Um número menor, no entanto, não é diagnosticado até os 18 anos ou mais. Esses pacientes geralmente têm uma forma mais branda da doença.

11
SINTOMAS: As manifestações clínicas típicas são tosse, diarreia crônica, e desnutrição. Entretanto, a doença pode se manifestar de outras maneiras, dependendo dos sistemas ou órgãos acometidos. Recém-nascidos podem incluir: Crescimento retardado Dificuldade para ganhar peso normalmente durante a infância Nenhuma evacuação nas primeiras 24 a 48 horas de vida Pele salgada

12
SINTOMAS: Funcionamento do intestino pode incluir:
Dor no abdome devido à constipação grave Aumento de gases, inchaço ou abdome que parece inchado (dilatado) Náusea e perda de apetite Fezes pálidas ou com cor de argila, odor fétido, têm muco ou flutuantes Perda de peso
 Náusea e perda de apetite. Fezes pálidas ou com cor de argila, odor fétido, têm muco ou flutuantes. Perda de peso.")
13
SINTOMAS: Os pulmões e seios nasais podem incluir:
Tosse ou aumento de muco nos seios nasais ou pulmões Fadiga Congestão nasal causada por pólipos nasais Febre Aumento da tosse Aumento na dificuldade para respirar Perda de apetite Aumento de escarro Dor ou pressão no seio nasal causada por infecção ou pólipos

14
SINTOMAS: Os sintomas podem ser observados em idade mais avançadas:
Infertilidade (em homens) Inflamação repetida do pâncreas (pancreatite) Sintomas respiratórios
 Inflamação repetida do pâncreas (pancreatite) Sintomas respiratórios.")
15
TRATAMENTO: Um diagnóstico precoce de FC e um plano de tratamento abrangente podem melhorar a sobrevivência e a qualidade de vida. O acompanhamento e monitoramento são muito importantes. O tratamento inclui: Antibióticos para prevenir e tratar de infecções nos pulmões e nos seios nasais. Medicamentos inalados para ajudar a abrir as vias respiratórias

16
TRATAMENTO: Terapia de substituição de enzima DNase para afinar o muco e facilitar a expectoração Alta concentração de soluções salinas (salina hipertônica) Vacina contra influenza e vacina antipneumocócica polissacarídica (PPV) O transplante de pulmão é uma opção em alguns casos A oxigenoterapia pode ser necessária à medida que a doença pulmonar piora Problemas pulmonares também são tratados com exercícios aeróbicos ou outras terapias para afinar o muco e facilitar a expectoração. Isso inclui coletes vibratórios, percussão torácica.
 Vacina contra influenza e vacina antipneumocócica polissacarídica (PPV) O transplante de pulmão é uma opção em alguns casos. A oxigenoterapia pode ser necessária à medida que a doença pulmonar piora. Problemas pulmonares também são tratados com exercícios aeróbicos ou outras terapias para afinar o muco e facilitar a expectoração. Isso inclui coletes vibratórios, percussão torácica.")
17
TRATAMENTO: Uma dieta especial rica em proteínas e calorias para crianças maiores e adultos Enzimas pancreáticas para ajudar a absorver gorduras e proteína Suplementos vitamínicos, especialmente vitaminas A, D, E e K Evitar o fumo, a poeira, a sujeira, as fumaças, os produtos químicos domésticos, a fumaça de lareira e o mofo ou bolor Limpar ou retirar muco ou secreções das vias respiratórias. Isso deve ser feito de uma a quatro vezes por dia. Beber líquidos em abundância.

18
INATIVIDADE FÍSICA: A habilidade para o exercício pode ser prejudicada pela presença de: - Doença pulmonar obstrutiva crônica (DPOC) - Desnutrição protéico energética (DPE). DIMINUIÇÃO DA MASSA MUSCULAR: A principal causa da diminuição da massa muscular é a desnutrição protéico-energética,causada pelo desequilíbrio entre a absorção e o requerimento de nutrientes ingeridos.
. DIMINUIÇÃO DA MASSA MUSCULAR: A principal causa da diminuição da massa muscular é a desnutrição protéico-energética,causada pelo desequilíbrio entre a absorção e o requerimento de nutrientes ingeridos.")
19
INATIVIDADE FÍSICA: DIMINUIÇÃO DA DENSIDADE MINERAL ÓSSEA (DMO): A perda óssea acelerada e, possivelmente a diminuição da formação óssea em pacientes com FC são moduladas, em parte, pelas citocinas – produzidas pelas células (macrófagos e neutrófilos) das vias aéreas - liberadas na infecção pulmonar. Outros fatores relacionados à DMO são: desnutrição, atraso puberal, deficiência de vitamina D, baixa ingestão e/ou absorção de cálcio, tratamento com corticosteroides, níveis diminuídos de hormônios sexuais e de insulina.
 das vias aéreas - liberadas na infecção pulmonar. Outros fatores relacionados à DMO são: desnutrição, atraso puberal, deficiência de vitamina D, baixa ingestão e/ou absorção de cálcio, tratamento com corticosteroides, níveis diminuídos de hormônios sexuais e de insulina.")
20
RELAÇÃO COM ATIVIDADE FÍSICA:
Para pacientes com FC são mais vantajosas as modalidades próximas ao local onde moram, e que envolvam resistência ao esforço: nadar, andar de bicicleta, passear, a ginástica e os jogos de rebatida (tênis, frescobol, ping-pong) são as mais indicadas, pois realizam grande mobilização do tórax. Já os jogos de bola (vôlei, futebol, basquete), transmitem experiências positivas e sociais, porém suas regras precisam ser alteradas. (KAMEL, 1994, apud KRÄMER, 2008). Brincadeiras como: subir em arvores; tiro ao alvo; jogo de taco; são exemplos de brincadeiras a serem realizadas.
 são as mais indicadas, pois realizam grande mobilização do tórax. Já os jogos de bola (vôlei, futebol, basquete), transmitem experiências positivas e sociais, porém suas regras precisam ser alteradas. (KAMEL, 1994, apud KRÄMER, 2008). Brincadeiras como: subir em arvores; tiro ao alvo; jogo de taco; são exemplos de brincadeiras a serem realizadas.")
21
FC X EXERCÍCIO AERÓBICO
Esses exercícios podem, seguramente, ser recomendados para pacientes com FC. Correr, caminhar, nadar, andar de bicicleta e jogar futebol são recomendados por no mínimo 20 minutos, pelo menos três vezes na semana para crianças e adolescentes com FC com comprometimento pulmonar de leve a moderado. Esses exercícios quando realizados de maneira regular, melhoram a capacidade cardiopulmonar, os níveis das atividades habituais, a tolerância ao exercício e a sensação de bem-estar de crianças sadias e com DPOC.

22
FC X EXERCÍCIO DE FORÇA Esses exercícios tem grande importância para a terapia dos pacientes com FC, porque a desnutrição e o processo inflamatório do pulmão levam à diminuição da massa corporal magra (hipotrofia muscular), apontada como principal fator da diminuição da capacidade para o exercício em indivíduos com FC. Aliado a esse quadro ocorre a perda de força da musculatura respiratória que causa hipercapnia (devido à ventilação pulmonar insuficiente) e aumenta ainda mais a limitação ao exercício. Desta maneira, aumentar a força da musculatura periférica e da musculatura respiratória é fundamental para melhorar a aptidão física.
, apontada como principal fator da diminuição da capacidade para o exercício em indivíduos com FC. Aliado a esse quadro ocorre a perda de força da musculatura respiratória que causa hipercapnia (devido à ventilação pulmonar insuficiente) e aumenta ainda mais a limitação ao exercício. Desta maneira, aumentar a força da musculatura periférica e da musculatura respiratória é fundamental para melhorar a aptidão física.")
23
RELATO DO ESTUDO DE CASO
Entrevista com uma adolescente portadora de fibrose cística, do sexo feminino, com idade de 22 anos, com estatura de 1,62 e peso de 54 Kg. Cabe ressaltar que no caso, a gravidade da doença era avaliada pelo médico como estado avançado. Como e quando foi detectado a FC? Quais os sintomas? Como é o tratamento? Pratica exercícios?

24
RELATO DESCRITIVO: A fibrocística tinha a doença de nascença, mas só foi descoberta aos 17 anos, ela relatou que nesta época chegou a pesar 33 kg e que estava muito desnutrida. Antes disso, era tratada como asma e bronquite. Foi nesta época em que teve a ajuda de um amigo que lhe pagou uma consulta particular e exames especiais com o Dr. Geraldo Richter. O Dr. Geraldo então se comoveu com o seu caso e disse que iria estudar e descobrir a doença. Para se ter certeza da doença foi feito vários exames como, por exemplo, o de suor que só era feito em Porto Alegre. Após a certeza de que a doença era fibrose cística iniciou-se rapidamente o tratamento.

25
RELATO DESCRITIVO: Encontra dificuldade nas atividades do dia a dia e para fazer exercícios, pois sente falta de ar, dor nas costas, dor no peito, dor nas pernas, tosse e catarro preso.

26
RELATO DESCRITIVO: Um exemplo de medicação é o creon pancreatina são 21 comprimidos ao dia. Enfim ela diz saber que todas as medicações são de extrema importância para controlar a sua doença. Tanto que às vezes o estado não manda e então ela tem que entrar no ministério público, porque é muito caro, daria em torno de R$ 2,000,00 reais ao mês. Além disso, ela diz ter várias vitaminas para tomar, auxiliando a deixá-la mais forte.

27
RELATO DESCRITIVO: A fibrose cística não tem cura, esta doença só pode ser tratada ou ser feito transplante. Mas neste caso estudado, houve demora de um doador de fígado, e a doença acabou se agravando e ela ficou muito fraca. Os médicos então acharam arriscado fazer transplante, e preferiram continuar apenas com o tratamento e acompanhamento médico. Os médicos recomendaram fazer fisioterapia, caminhar e andar de bicicleta.

28
CONCLUSÃO: Após a realização deste estudo foi possível concluir que FC é uma doença pouco conhecida , a qual afeta muitos órgãos e exige um tratamento baseado em muitos medicamentos. Destacamos também que a atividade física é importante e benéfica para uma melhor qualidade de vida, pois fibrose cística não tem cura. Observamos no estudo de caso que as atividades físicas que ela prática mais assiduamente são ciclismo e caminhada, e reconhece que após esses exercícios sente-se melhor. Os exercícios ajudam a soltar o muco, reduzem a respiração curta, deteorização dos músculos. Mas que encontra dificuldade nas atividades do dia a dia e para fazer exercícios, pois sente falta de ar, dor nas costas, dor no peito, dor nas pernas, tosse e catarro preso.

29
REFERÊNCIAS: http://www.scielo.br/pdf/jbpneu/v33n2/08.pdf
Análise comparativa e reprodutibilidade do teste de caminhada com carga progressiva (modificado) em crianças normais e em portadoras de fibrose cística Exercício aeróbico, treinamento de força muscular e testes de aptidão física para adolescentes com fibrose cística: revisão da literatura Rocha, Kátia Bones, Moreira, Mariana Calesso, de Oliveira,Viviane Ziebell -Adolescência em pacientes portadores de fibrose cística. Aletheia 20, jul./dez. 2004 Krämer, Diego Rafael – 2008, O IMPACTO DA ATIVIDADE FÍSICA PARA O PORTADOR DE FIBROSE CÍSTICA: Um estudo de caso na zona rural de Venâncio Aires
 em crianças normais e em portadoras de fibrose cística. Exercício aeróbico, treinamento de força muscular e testes de aptidão física para adolescentes com fibrose cística: revisão da literatura. Rocha, Kátia Bones, Moreira, Mariana Calesso, de Oliveira,Viviane Ziebell -Adolescência em pacientes portadores de fibrose cística. Aletheia 20, jul./dez Krämer, Diego Rafael – 2008, O IMPACTO DA ATIVIDADE FÍSICA PARA O PORTADOR DE FIBROSE CÍSTICA: Um estudo de caso na zona rural de Venâncio Aires.")
Apresentações semelhantes










 ou mucoviscidose é uma doença autossômica recessiva caracterizada por secreções anormalmente densas das glândulas mucosas,>")








