Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Genética de doenças Alexandre Ferreira Bilhalva
UNIVERSIDADE FEDERAL DE PELOTAS GRADUAÇÃO EM BIOTECNOLOGIA DISCIPLINA DE GENÔMICA II Genética de doenças Alexandre Ferreira Bilhalva Arthur de Siqueira Brahm Henrique Ramos Angelo Profª Fabiana Seixas, Dra.

2
Introdução Histórico: Hereditariedade – Mendel (1856)
Melhoramento Genético Século XX

3
Introdução Sequenciamento do genoma humano!!
Fatores genéticos de doenças!

4
Introdução Genética Molecular Citogenética Genética Bioquímica
Genética fisiológica

5
Função dos genes envolvidos
Tratamento, prevenção Hereditariedade Diagnósticos genéticos preventivos Diagnóstico de doenças

6
Descreve as FUNÇÕES dos genes.
– - - Estuda a natureza física e o funcionamento do material genético contido no conjunto de cromossomos de cada espécie. Determina a posição cromossômica e a sequência de bases dos genes de diversas espécies. Descreve as FUNÇÕES dos genes. Analisa como os genes estão sendo expressos, ou silenciados. Genômica Genômica Estrutural Genômica Funcional

7
Síndrome de Tourette Transtorno genético neuropsiquiátrico Infância
Tiques motores e vocais crônicos 1/100 é afetada pela ST

8
Histórico Jean Marc Gaspard Itard – 1825 – Marquesa de Dampierre
Gilles de la Tourette – 1885 – “Estudo de uma enfermidade nervosa”

9
Sintomas Normalmente entre 6 e 7 anos.
Obrigatórios tiques motores, e ao menos um vocal.

11
Patofisiologia Tiques e movimentos involuntários
Tálamo, Gânglios basais, Córtex Frontal

12
Artigo

13
Genética Genes Slit e Trk-like 1 (SLITRK1)
Proteína transmembrana rica em Leucina Inversão no cromossomo 13 (q31.1)
")
14
Genética Mapeamento por Fish

15
Genética Wild-type SLITRK1: Mutante SLITRK1:

16
Genética SLITRK1 – diversas regiões do cérebro de um feto.
Teste em camundongos – Wild-type SLITRK1 x SLITRK1 Mutante Diferença no tamanho dos dentritos

17
Resultados

18
Tratamento Não há cura Depende da severidade dos sintomas
Terapias comportamentais; uso de fármacos Tratamento de tiques e outros sintomas

19
Terapias -> isolamento social; depressão
Tratamento Terapias -> isolamento social; depressão

20
Fármacos mais eficientes(tiques):
Tratamento Fármacos mais eficientes(tiques): Neurolépticos Antidepressivos
: Neurolépticos. Antidepressivos.")
21
Bloqueadores dos receptores D2
Tratamento Bloqueadores dos receptores D2 DOPAMINA - SNC

22
Associações National Tourette Syndrome Association (EUA).
Associação Brasileira de Síndrome de Tourette, Tiques e Transtorno Obsessivo Compulsivo . National Tourette Syndrome Association (EUA).
.")
23
Doença de Huntington

24
Doença de Huntington Doença neurodegenerativa genética.
A ocorrência dela depende da Etnia, e de padrões de migração populacional.

25
Doença de Huntington Causada por uma mutação autossômica dominante.
Um pai com um alelo: 50% Dois pais com um alelo: 75% Qualquer pai com dois alelos: 100%

26
Sintomas Sintomas geralmente aparecem entre anos, movimentos involuntários, bruscos e irregulares. Perda de função motora. Perda da visão periférica. Dificuldade para comer com as mãos, mastigar e deglutir. Dilaceração dos nervos.

27
Regiões Danificadas Afeta os Neurônios do corpo estriado e do córtex. Causando a morte dos mesmos.

28
Genética Autossomica dominante, cromossomo 4, região IT15.
Onde uma sequencia CAG é repetida varias vezes, de forma a codificar para a proteina mutada.

29
Genética CAG é o código genético do aminoácido Glutamina.
Esta seqüência repetida, é conhecida como Mutação Poliglutamica. Os afetados possuem mais de 36 repetições da sequencia CAG.

30
Huntingtina 6-35 resíduos de glutamina Principalmente no cérebro
Citoplasma Expressa principalmente no cérebro e nos testiculos Não tem sequencia homologa com outras proteinas Ausência causa morte em camundongos

31
Huntingtina Essencial ao desenvolvimento
Associadas a microtúbulos e vesículas Sinalização Transporte de materiais Ligação de proteínas e outras estruturas Apoptose Indispensável para o desenvolvimento pré-natal

32
A penetrância completa
Huntingtina Mutante >36 Glutaminas Expansão do Exon I Apresenta graus de severidade dependendo da quantidade de repetições de glutamina. Núcleo Alterações no sistema de apoptose e exocitose de substancias pelos neurônios Repetir a contagem Classificação Estado da doença <28 Normal Não afetado 28-35 Intermediário 36-40 Penetrância reduzida + / - Afetados > 40 A penetrância completa Afetado

33
Alterações Celulares devido a Huntingtina mutacionada
A natureza polar da glutamina, causa interações com outras proteínas huntingtina

34
Alterações Celulares devido a Huntingtina mutacionada
Esses agregados se acumulam formando corpos de inclusão dentro da célula, causando grande interferência no funcionamento dos neurônios.

35
Alterações Celulares devido a Huntingtina mutacionada
Essa interferência ocorre pois os corpos de inclusão dificultam o movimento das vesículas de neurotransmissores dentro do axônio, reduzindo a sinalização dos neurônios.

36
Alterações Celulares devido a Huntingtina mutacionada
A huntingtina mutacionada pode também causar efeito nas proteínas chaperone que ajudam no dobramento correto de proteínas

37
Alterações Celulares devido a Huntingtina mutacionada
Esses efeitos são aumentados com a interação com a proteína Rhes, que causa o desagregamento da huntingtina mutacionada, gerando uma forma mais toxicológica da proteína.

38
Alterações Celulares devido a Huntingtina mutacionada
Também causa dano as mitocôndria das células estriadas, e aumenta a vulnerabilidade a glutamina, esta causa uma estimulação em excesso a neurotoxinas causando morte celular.

39
Alterações Celulares devido a Huntingtina mutacionada
Os agregados de huntingtina são clivados em pequenos pedaços, que entram no núcleo do neurônio e atrapalham a transcrição de outras proteínas causando a morte celular.

40
Diagnóstico imagético
Tomografia Computadorizada Ressonância Magnética

41
Diagnóstico genético Confirmação da doença de huntington;
Melhor método para realizar diagnóstico diferencial;

42
Teste Genético em pacientes Sintomaticos
Sintomas neurológicos compatíveis com DH; Diagnóstico diferencial de outras doenças Se o paciente apresentar a expansão do trinucleotídeo e não tiver história familiar positiva, deve-se verificar a paternidade.

43
Teste genético em pacientes assintomáticos
Filhos de pais portadores de DH; 50% de chance Planejamento pessoal e familiar; Alterações psiquiátricas

44
Diagnóstico pré-natal
Cordocentese: Sangue Fetal Amniocentese: Liquido Aminiotico

45
Tratamento com RNAi

46
Doença de Alzheimer

47
Doença de Alzheimer Forma de demência mais comum no mundo
Relação entre idade e demência Deteriora progressivamente

48
Doença de Alzheimer Não possui cura Descrita em 1901, Alois Alzheimer

49
Novos afetados a cada mil/ por ano
Doença de Alzheimer Idade Novos afetados a cada mil/ por ano 65–69 3 70–74 6 75–79 9 80–84 23 85–89 40 90– 69

50
Sintomas – Pré-demência
Raramente são notados Confundidos com outros problemas Problemas sutis

51
Sintomas - Leves Queda na memória e no aprendizado
Problemas relacionados à linguística Deficiências motoras em atividades finas

52
Sintomas - Moderados Problemas linguísticos ficam evidentes
Atividade motora se torna altamente deficiente Mudanças de humor e comportamento erradico

53
Sintomas - Graves Completamente dependente de outros
Capacidade linguística quase nula Atividade motora deteriorada com apatia e exaustão

54
Fisiopatologia Ocorre a perda de neurônios e sinapses no córtex e região subcortical. Tantos amiloides e neurofibrilas são visíveis por microscopia. Corpos de Lewy também são visíveis.

55
Diagnóstico Entrevista clínica Aparelhos de imagens

56
Causa Não se sabe ao certo. Envelhecimento e stress oxidativo possuem um papel importante. Principais teorias:

57
Causa - Acetilcolinergica

58
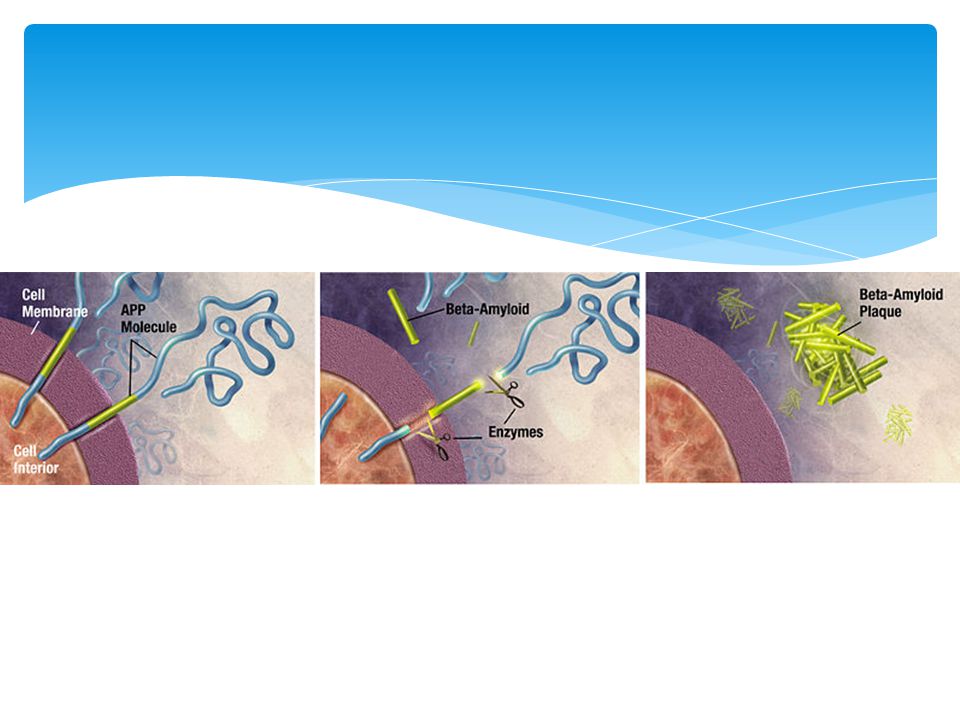
Causa – Beta amiloide

60
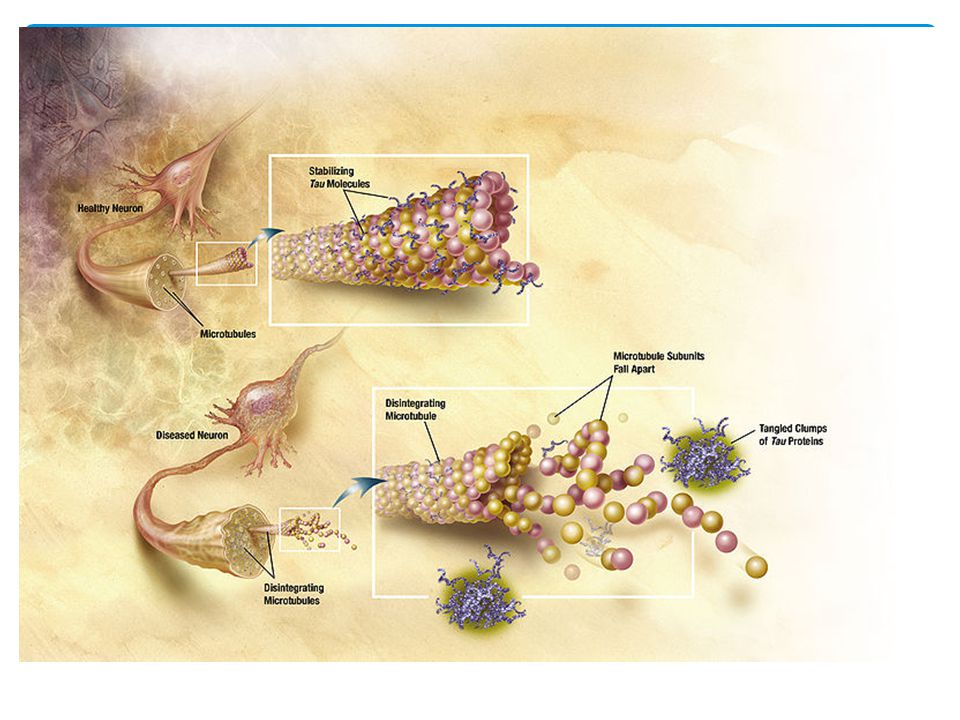
Causa - Neurofibrilas

62
Genética Manifestação esporádica, mas apresenta genes de risco.
0,1 % dos casos apresentam características autossômicas dominantes (DAF).
.")
63
Genética Genes importantes na DAF: - Proteína precursora de amilóide
- Presenilina 1 - Presenilina 2

64
Genética Genes de fatores de risco: - Épsilon 4 alelo
- APOE (40-80% apresentam) Outros genes possuem papel regulador. EX: Populações nigerianas.
 Outros genes possuem papel regulador. EX: Populações nigerianas.")
65
Tratamento Fármacos principais:
4 inibidores da acetilcolinaesterase (Tacrina, Rivastigamina, Galantamina e Donepezil). 1 antagonista do receptor de NMDAR (Memantina).
. 1 antagonista do receptor de NMDAR (Memantina).")
66
Tratamento Antipsicóticos para sintomas mentais.
Tratamento para problemas psicológicos.

67
Prevenção Não se conhece mecanismos concretos
Dieta do Mediterrâneo é eficiente Atividade cognitiva constante

68
Futuro

69
Obrigado!

70
Bibliografia http://en.wikipedia.org/wiki/Alzheimer%27s_disease

Apresentações semelhantes