Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Intolerância congênita aos carboidratos
Lanna Beatrice T. Maluf, M.D.* & Ulysses Fagundes Neto, M.D., Ph.D.** *Especializanda em Gastroenterologia Pediátrica ** Professor Titular Disciplina de Gastroenterologia Departamento de Pediatria Escola Paulista de Medicina UNIFESP

2
Introdução Os carboidratos são as biomoléculas mais abundantes na natureza, e apresentam a seguinte fórmula geral: [C(H2O)]n, daí o nome carboidratos ou hidratos de carbono. Desempenham uma ampla variedade de funções, tais como: Fonte de energia – representam de 40 a 50% da ingestão energética diária Reserva de energia Matéria prima para a biossíntese de outras macromoléculas Evans PR. et al. Scand J Gastroenterol 1998; 33:
]n, daí o nome carboidratos ou hidratos de carbono. Desempenham uma ampla variedade de funções, tais como: Fonte de energia – representam de 40 a 50% da ingestão energética diária. Reserva de energia. Matéria prima para a biossíntese de outras macromoléculas. Evans PR. et al. Scand J Gastroenterol 1998; 33:")
3
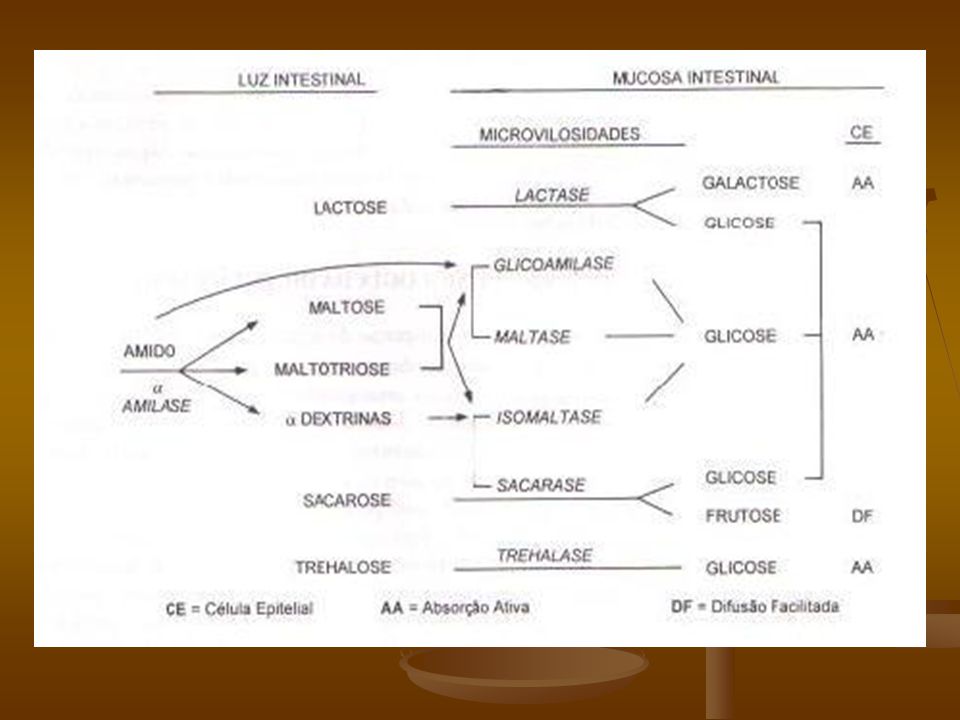
Os carboidratos podem ser consumidos nas formas mais variadas, desde simples moléculas como a glicose até os não absorvíveis como as fibras insolúveis.(1) Classificação: A) Monossacarídeos - açúcar simples Tipos: glicose, galactose, frutose B) Dissacarídeos – glicosídeos (formados a partir da ligação de 2 monossacarídeos através de ligações especiais denominadas ligações glicosídicas). Tipos: sacarose, maltose, lactose (1) Aggett PJ. et al. J Pediatr Gastroenterol Nutr 2003; 36:
 Monossacarídeos - açúcar simples. Tipos: glicose, galactose, frutose. B) Dissacarídeos – glicosídeos (formados a partir da ligação de 2 monossacarídeos através de ligações especiais denominadas ligações glicosídicas). Tipos: sacarose, maltose, lactose. (1) Aggett PJ. et al. J Pediatr Gastroenterol Nutr 2003; 36:")
4
Tipos: amido, glicogênio, celulose.
C) Polissacarídeos - carboidratos complexos, macromoléculas formadas por milhares de unidades monossacarídicas ligadas entre si por ligações glicosídicas. Funções biológicas: armazenadora de combustível; elementos estruturais. Tipos: amido, glicogênio, celulose. D) Oligossacarídeos: dextrinomaltoses As moléculas maiores como os di, oligo e polissacarídeos necessitam ser quebradas, através da ação de enzimas, até a forma de monossacarídeos para haver absorção. Gudmand – Hoyer et al. Scand J Gastroenterol 1996; 31 suppl 216: Curr Opin Gatroenterol; 20(2):162-7; 2004 Mar.
 Polissacarídeos - carboidratos complexos, macromoléculas formadas por milhares de unidades monossacarídicas ligadas entre si por ligações glicosídicas. Funções biológicas: armazenadora de combustível; elementos estruturais. Tipos: amido, glicogênio, celulose. D) Oligossacarídeos: dextrinomaltoses. As moléculas maiores como os di, oligo e polissacarídeos necessitam ser quebradas, através da ação de enzimas, até a forma de monossacarídeos para haver absorção. Gudmand – Hoyer et al. Scand J Gastroenterol 1996; 31 suppl 216: Curr Opin Gatroenterol; 20(2):162-7; 2004 Mar.")
6
INTOLERÂNCIA AOS CARBOIDRATOS
Intolerância aos carboidratos é uma denominação utilizada para caracterizar manifestações clínicas que ocorrem devido a alterações da digestão e/ou da absorção dos mesmos.(1) As alterações primárias ocorrem por deficiências congênitas de transportadores de monossacarídeos ou de enzimas que hidrolisam os açúcares mais complexos, portanto, essas alterações podem ocorrer através da ausência completa ou por deficiência de atividade dos diversos complexos enzimáticos envolvidos na digestão dos carboidratos.(2) (1) Curr Opin Gatroenterol; 20(2):162-7; 2004 Mar. (2) Arola H. Scand J Gastroenterol 1994, 29: Hertzler SR et al. Am J Clin Nutr 1996; 64:
 As alterações primárias ocorrem por deficiências congênitas de transportadores de monossacarídeos ou de enzimas que hidrolisam os açúcares mais complexos, portanto, essas alterações podem ocorrer através da ausência completa ou por deficiência de atividade dos diversos complexos enzimáticos envolvidos na digestão dos carboidratos.(2) (1) Curr Opin Gatroenterol; 20(2):162-7; 2004 Mar. (2) Arola H. Scand J Gastroenterol 1994, 29: Hertzler SR et al. Am J Clin Nutr 1996; 64:")
7
Classificação 1) Intolerância aos polissacarídeos Intolerância ao amido 2) Intolerância aos dissacarídeos Intolerância à lactose Deficiência congênita de lactase Deficiência ontogenética de lactase ou hipolactasia tipo adulto Deficiência de lactase adquirida ou intolerância secundária à lactose Alactasia congênita

8
Intolerância à sacarose-isomaltose
Deficiência congênita de sacarase-isomaltase 3) Intolerância a monossacarídeos Má absorção primária de glicose-galactose Intolerância hereditária à frutose Payne ML et al. J Am Diet Assoc 1997(5): Aggett P J. J Pediatr Gastroenterol Nutr 2003; 36:329-37
 Intolerância a monossacarídeos. Má absorção primária de glicose-galactose. Intolerância hereditária à frutose. Payne ML et al. J Am Diet Assoc 1997(5): Aggett P J. J Pediatr Gastroenterol Nutr 2003; 36:")
9
I) Intolerância à lactose
A lactase (complexo enzimático lactase-florizina-hidrolase), está localizada, preferencialmente no topo das vilosidades, nas microvilosidades dos enterócitos maduros em todo o intestino delgado e sua atividade não sofre influência da quantidade de lactose ingerida bem como com relação a outros açúcares da dieta. A função da lactase é degradar a lactose em seus dois monossacarídeos constituintes, a saber: glicose e galactose. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition.
, está localizada, preferencialmente no topo das vilosidades, nas microvilosidades dos enterócitos maduros em todo o intestino delgado e sua atividade não sofre influência da quantidade de lactose ingerida bem como com relação a outros açúcares da dieta. A função da lactase é degradar a lactose em seus dois monossacarídeos constituintes, a saber: glicose e galactose. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition.")
10
O gene que controla sua produção está localizado no braço longo do cromossomo 2 (2q21).
Na maioria da população o padrão da atividade lactásica ao longo do desenvolvimento é caracterizado por um aumento durante o período fetal tardio (35-38sem), que prossegue até elevados níveis por volta do nascimento. A produção de lactase declina na infância tardia, alcançando nível estacionário na idade pré escolar e baixos níveis de atividade na idade adulta. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition. Evans PR. et al. Scand J Gastroenterol 1998; 33:
, que prossegue até elevados níveis por volta do nascimento. A produção de lactase declina na infância tardia, alcançando nível estacionário na idade pré escolar e baixos níveis de atividade na idade adulta. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition. Evans PR. et al. Scand J Gastroenterol 1998; 33:")
11
I-A) Deficiência congênita de lactase
Foi descrita pela 1ª vez por Holzel et al., em 1959, na Finlândia, também denominada alactasia congênita. Trata-se de uma anormalidade genética muito rara, herdada por mecanismo autossômico recessivo. Qadro Clínico: Manifestação no recém nascido logo após a 1ª ou 2ª ingestão de leite humano ou animal. Distensão e dor abdominal, excessiva eliminação de flatos, diarréia líquida, volumosa, vômitos, desidratação e acidose metabólica. Dermatite perianal, parada do crescimento e desnutrição. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition Hozel et al. Lancet 1959; i: Arch.Dis.Chid 58:246-52, 1983

12
Critérios diagnósticos para deficiência congênita:
a) ausência de atividade enzimática desde o nascimento e persistência por toda a vida. b) ausência de intolerância a outro mono e/ou dissacarídeo. c) morfologia normal da mucosa do intestino delgado. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition
 ausência de atividade enzimática desde o nascimento e persistência por toda a vida. b) ausência de intolerância a outro mono e/ou dissacarídeo. c) morfologia normal da mucosa do intestino delgado. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition.")
13
I-B) Deficiência Ontogenética de Lactase
Sinonímia: Hipolactasia Primária do Adulto, Deficiência de Lactase do Adulto. É a forma mais comum de deficiência genética. A prevalência é elevada entre árabes, habitantes do sul da Itália, esquimós, negros americanos, índios, orientais, africanos e provavelmente brasileiros. Evans PR. et al. Scand J Gastroenterol 1998; 33: .Aggett P J. J Pediatr Gastroenterol Nutr 2003; 36:

14
Manifestações clínicas são menos intensas e mais tardias, as quais são idade dependente.
A diminuição da atividade enzimática começa aos 5 anos em crianças brancas e 3 anos em negras, ou mesmo, pode ocorrer só na vida adulta em alguns casos. É rara a ausência total de atividade enzimática, a qual persiste em aproximadamente 10% do seu valor máximo. A criança pode ser assintomática, caracterizando-se como mau absorvedor, ou referir que não gosta de leite, recusando-o, o que pode mascarar a intolerância. Treem WR. J Pediatr Gastroenterol Nutr 1999; 28:

15
Distensão abdominal, flatulência e dor abdominal recorrente, em cólica espástica, periumbilical ou difusa no abdome, de intensidade variável, sendo maior com o aumento da ingestão de leite e/ou derivados são os sintomas mais frequentes. Tem sido apontada como causa de Síndrome do Intestino Irritável em adultos e Dor Abdominal Cônica Recorrente em crianças. É importante investigar outros casos de intolerância na família, principalmente nos pais. Treem WR. J pediatr Gastroenterol Nutr 1999; 28: Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition

16
Forte suspeita clínica. Diagnóstico laboratorial:
1- determinação do pH e pesquisa de substâncias redutoras nas fezes; 2- teste de tolerância com curva glicêmica e/ou teste do Hidrogênio no ar expirado. Padrão ouro: determinação da atividade enzimática em fragmento de mucosa intestinal e estudo imuno-histoquímico, na presença de mucosa de intestino delgado histologicamente normal. Arola H. Scand J. Gastroenterol 1994;29 suppl 202:26-35 Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition

17
Tratamento Consiste na retirada do dissacarídeo da dieta, uma vez que o fenômeno de indução de atividade da enzima pela dieta não existe. Orientar bem a família – problema de base. Deficiência congênita de lactase – dieta isenta de lactose. Deficiência ontogenética – redução na quantidade de leite e derivados de acordo com a tolerância individual. Administração exógena de Lactase J Pediatr Gastroenterol Nutr ;35(4):573-9,2002 Oct
:573-9,2002 Oct.")
18
II) Intolerância a sacarose- isomaltose
O complexo enzimático sacarase-isomaltase é responsável por toda atividade da sacarase, praticamente toda atividade da isomaltase e, aproximadamente 80% da maltase. Esse complexo também se distribui por todo o intestino delgado, porém em proporções decrescentes no íleo. É a 2ª deficiência primária das dissacaridases mais freqüentes na espécie humana com prevalência entre 0,1 e 5%. Karnsakul W et al. J Pediatr Gastroenterol Nutr 2002;35 ;551-6

19
Sua descrição foi em princípio feita em esquimós da Groenlândia, Canadá e Índios Canadenses.
Esse complexo enzimático forma-se dentro do enterócito e é translocado para o reticulo endoplasmático e aparelho de Golgi, onde é glicosilado e transportado até a membrana da célula, sendo então clivado e transformado em sacarase e isomaltase por ação das peptidases pancreáticas presentes na luz intestinal. Nichols BL et al. J Pediatr Gastroenterol Nutr 2002;35:573-9

20
Pode ser diagnosticado em bebês, crianças ou adultos.
O gene codificador desse complexo está no braço do cromossomo 3 (3q22-q26), tendo como forma mais freqüente a substituição de prolina por glutamina na sub-unidade sacarase, sendo transmitido através de caráter autossômico recessivo. Pode ser diagnosticado em bebês, crianças ou adultos. Heterozigotos podem ter atividade intermediária da enzima. Jacob R et al. J Clin Invest 2000; 106(2):281-7 Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition
, tendo como forma mais freqüente a substituição de prolina por glutamina na sub-unidade sacarase, sendo transmitido através de caráter autossômico recessivo. Pode ser diagnosticado em bebês, crianças ou adultos. Heterozigotos podem ter atividade intermediária da enzima. Jacob R et al. J Clin Invest 2000; 106(2): Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition.")
21
São 5 os tipos de defeitos moleculares responsáveis por essa deficiência:
Tipo 1) O defeito está no aparelho de Golgi, com atividade de sacarase ausente e da isomaltase reduzida. Tipo 2) Defeito no Retículo Endoplásmico Rugoso, com atividade da sacarase e da isomaltase ausente. Tipo 3) Defeito na vilosidade por alteração no ponto catalítico da sacarase, (atividade ausente) e da isomaltase (atividade normal).
 O defeito está no aparelho de Golgi, com atividade de sacarase ausente e da isomaltase reduzida. Tipo 2) Defeito no Retículo Endoplásmico Rugoso, com atividade da sacarase e da isomaltase ausente. Tipo 3) Defeito na vilosidade por alteração no ponto catalítico da sacarase, (atividade ausente) e da isomaltase (atividade normal).")
22
Tipo 4) Idem ao tipo 3, porém com degradação intracelular da sacarase.
Tipo 5) O complexo enzimático permanece no RER sem sofrer glicosilação, com atividade ausente de ambas enzimas. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition
 O complexo enzimático permanece no RER sem sofrer glicosilação, com atividade ausente de ambas enzimas. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition.")
23
QUADRO CLÍNICO Apresentação clínica é variável e ocorre, provavelmente, devido os diversos tipos de defeito tendo início após a introdução desses açúcares na dieta da criança. Diarréia é o sinal mais freqüente e pode ser intermitente, volumosa, líquida, acompanhada de distensão abdominal, eventualmente vômitos, má absorção e dificuldade para crescer.

24
Nas crianças, porém, pode ser detectado durante a evolução para diarréia crônica, sem retardo de crescimento. Alternativamente pode não se manifestar até a idade adulta, quando os pacientes se queixam de distensão abdominal intermitente, flatulência e diarréia - sintomas semelhantes à Síndrome do Intestino Irritável. Karnsakul W et al. J Pediatr Gastroenterol Nutr 2002;35 ;551-6 Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition BMC Pediatr;2:4, 2002 Apr 25

25
Diagnóstico Padrão ouro: determinação da atividade enzimática na mucosa intestinal determinada em material de Biópsia de Intestino Delgado. Laboratorial: determinação do pH fecal <4 ausência de substâncias redutoras nas fezes curva glicêmica ou teste de H2 no ar expirado (usando sacarose como substrato). Nichols BL et al. J Pediatr Gastroenterol Nutr 2002; 30:
. Nichols BL et al. J Pediatr Gastroenterol Nutr 2002; 30:")
26
Tratamento Consiste na retirada da sacarose da dieta e, em casos raros, do amido (fonte de isomaltose). O açúcar da cana pode ser substituído por lactose, glicose e/ou frutose. Algumas frutas contém pouca sacarose e podem ser testadas, quanto a tolerância, em pequeno volume por vez: mamão papaia, figo, morango, tâmara seca, caqui, goiaba, pêra, maracujá. Abacate, uva e romã - não contém sacarose.

27
Dentre as hortaliças devem ser evitadas, as que contém maior teor de sacarose, tais como: beterraba, batata doce, ervilha, cenoura e cebola. Desenvolveu-se uma solução de sacarase produzida por Saccharomyces cerevisiae capaz de exercer efeito na hidrólise da sacarose em intestino de portadores dessa deficiência. Essa solução não substitui a enzima mas diminui a restrição dietética a que esses pacientes são submetidos. Treem W et al. J Pediatr Gastroenterol Nutr 1999;28: Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition

28
INTOLERÂNCIA AOS MONOSSACARÍDEOS
I) Má absorção congênita de glicose-galactose O estudo da intolerância dos monossacarídeos teve início em 1962 com Lindquist et al, e foi descrita pela 1ª vez por Burke et al. É uma síndrome diarréica familiar de herança autossômica recessiva, que cursa com diarréia de início precoce.
 Má absorção congênita de glicose-galactose. O estudo da intolerância dos monossacarídeos teve início em 1962 com Lindquist et al, e foi descrita pela 1ª vez por Burke et al. É uma síndrome diarréica familiar de herança autossômica recessiva, que cursa com diarréia de início precoce.")
29
É caracterizada por um defeito primário de absorção de 2 monossacarídeos, os quais são transportados através da membrana por um carregador comum tal como o sódio. O carregador localiza-se no bordo em escova do enterócito, possuindo afinidade e receptores para glicose e galactose. A presença de sódio é importante para a afinidade da glicose, cujo transporte é realizado com gasto de energia. Abdullah AMA et al.J Pediatr Gastroenterol Nutr 1996;23:561-64

30
O sódio absorvido dentro da célula é bombeado ativamente para fora da mesma à custa da bomba Na-K-ATPase, através da membrana basolateral. O monossacarídeo poderá ser utilizado pela célula ou transportado passivamente para a circulação. O gene responsável pelo não funcionamento do transporte de glicose e galactose na borda em escova dos enterócitos situa-se no braço longo do cromossomo 22(22q13.1). Evans L et al.J Pediatr Gastroenterol Nutr 1985;4:878-86 Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition Smith W. Pediatric
. Evans L et al.J Pediatr Gastroenterol Nutr 1985;4: Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition. Smith W. Pediatric.")
31
Vômitos são freqüentes.
QUADRO CLÍNICO Diarréia que tem início após entre os primeiros dias de vida até 2 semanas após o nascimento, caracterizada por fezes líquidas, explosivas, distensão abdominal e desidratação grave. Vômitos são freqüentes. Pode ocorrer glicosúria transitória ou permanente. BMC Pediatr;2:4,2002 Apr 25 Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition

32
Diagnóstico Teste de sobrecarga oral de glicose, galactose, lactose (0,5g/kg), com curvas achatadas de glicose no sangue, como resultado da falta de absorção intestinal da glicose e galactose. Teste de H2 no ar expirado. Fezes com pH ácido e com grande quantidade de substâncias redutoras. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition
, com curvas achatadas de glicose no sangue, como resultado da falta de absorção intestinal da glicose e galactose. Teste de H2 no ar expirado. Fezes com pH ácido e com grande quantidade de substâncias redutoras. Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition.")
33
Tratamento Em princípio, visa medidas de suporte, posto que o paciente pode apresentar desidratação grave. Glicose e galactose devem ser excluídas da dieta, nesta fase os HC devem ser fornecidos por via intravenosa. Como a frutose é o único carboidrato tolerado sua adição deve ser iniciada prontamente. Posteriormente, entre 6 e 8mêses de vida, alimentos amiláceos e pequenas quantidades de sacarose podem ser utilizados de acordo com a tolerância demonstrada pelo paciente.

34
Aqueles pacientes que conseguem sobreviver além dos 4 anos de idade, tem sido observada grande aumento da tolerância aos monossacarídeos (glicose e galactose). Na maioria dos casos, o crescimento e o desenvolvimento são normais quando a glicose e a galactose são retirados da dieta precocemente. Maffei H et al. Doenças gastroenterológicas em Pediatria.1 ed, 1996:157-65 Smith W. Pediatric Gastrointestinal Disease 2004; 43: , fourth edition

35
II) Galactosemia A galactosemia é uma doença rara do metabolismo da galactose. A alteração se encontra em uma das enzimas responsáveis pela conversão da galactose em glicose e dependendo da enzima defeituosa, a doença se manifesta de maneira diferente. Classificação: Galactosemia tipo 1, 2 e a tipo 3. Smith W. Pediatric Gastrointestinal Disease 2004; 55: , fourth edition

36
Patogenia A lactose, que é o principal carboidrato do leite é um dissacarídeo que contém galactose e glicose. Quando ingerida é hidrolisada pela lactase intestinal. Em condições normais, a galactose absorvida é convertida em glicose no fígado. A 1ª reação nesta via é a fosforilação da galactose em galactose-1-fosfato pela galactoquinase (especificado por um gene no cromossomo 17).
.")
37
Galactose + ATP galactoquinase galactose-1-fosfato.
A etapa seguinte envolve a conversão da gactose-1-fosfato em glicose-1-fosfato pela GALT (cujo gene reside no cromossomo 9) Galactose-1-fosfato + UDP-glicose GALT UDP galactose + glicose-1-fosfato. Os açúcares difosfato de uridina (UDP) podem sofrer interconversão reversível através de uma reação catalisada por uma epimerase (UDP-galactose-4-epimerase). UDP-galactose UDP-glicose. Mol Genet Metab;86(3):360-71,2005Nov Biochem Biophys Res Commun;271(2): ,2000 May
 Galactose-1-fosfato + UDP-glicose GALT UDP galactose + glicose-1-fosfato. Os açúcares difosfato de uridina (UDP) podem sofrer interconversão reversível através de uma reação catalisada por uma epimerase (UDP-galactose-4-epimerase). UDP-galactose UDP-glicose. Mol Genet Metab;86(3):360-71,2005Nov. Biochem Biophys Res Commun;271(2): ,2000 May.")
38
A) Galactosemia tipo 1 É a deficiência no metabolismo da galactose por deficiência da enzima galactose-1-fosfato uridil transferase (GALT). A patologia se traduz no defeito genético da enzima GALT, o que gera acúmulo de galactose-1-fosfato e galactose tecidual. A incidência é de 1: na população branca e a idade alvo é preferencialmente neonatos.

39
O fator de risco dessa doença é a presença do gene recessivo para a galactosemia nos pais. O gene da GALT se encontra no cromossomo 9p13, e a probabilidade da criança nascer com a deficiência é igual tanto para meninos e como para meninas. Os danos podem se iniciar na fase pré natal a partir da galactose transplacentária vinda da mãe heterozigota. Hum Mutat;23(4):396,2004 Apr J Inherit Metab Dis;25(8):629-34,2002 Dec
:396,2004 Apr. J Inherit Metab Dis;25(8):629-34,2002 Dec.")
40
As características clínicas da doença podem se apresentar de diversas maneiras diferentes e surgem poucos dias a semanas após o nascimento: A) As principais manifestações hepáticas e gastrointestinais são: irritabilidade, letargia, vômitos, dificuldade de alimentação, baixo ganho de peso, hepatoesplenomegalia, ascite, cirrose hepática e outras complicações. B) A Síndrome de Fanconi: vômitos, desidratação, fraqueza e febre inexplicada, anorexia, constipação, polidipsia, poliuria e distúrbios do crescimento.
 As principais manifestações hepáticas e gastrointestinais são: irritabilidade, letargia, vômitos, dificuldade de alimentação, baixo ganho de peso, hepatoesplenomegalia, ascite, cirrose hepática e outras complicações. B) A Síndrome de Fanconi: vômitos, desidratação, fraqueza e febre inexplicada, anorexia, constipação, polidipsia, poliuria e distúrbios do crescimento.")
41
C) Outras manifestações:
neurológicas: retardo mental (irreversível), problemas da fala e coordenação motora. endócrinas: disfunção ovariana com amenorréia 1ª ou 2ª, provável aumento de risco de câncer ovariano. oculares: catarata, no cristalino a galactose é convertida pela aldolase redutase em galactitol, um açúcar ao qual o cristalino é impermeável, e em conseqüência ocorre hidratação excessiva, redução do glutation no cristalino e formação de cataratas. infecciosas. J Inherit Metab Dis;25(8):629-34,2002 Dec J Trop Pediatr;47(6):372-3,2001Dec
, problemas da fala e coordenação motora. endócrinas: disfunção ovariana com amenorréia 1ª ou 2ª, provável aumento de risco de câncer ovariano. oculares: catarata, no cristalino a galactose é convertida pela aldolase redutase em galactitol, um açúcar ao qual o cristalino é impermeável, e em conseqüência ocorre hidratação excessiva, redução do glutation no cristalino e formação de cataratas. infecciosas. J Inherit Metab Dis;25(8):629-34,2002 Dec. J Trop Pediatr;47(6):372-3,2001Dec.")
42
B) Galactosemia tipo 2 É a disfunção do metabolismo da galactose pela deficiência da atividade da galactoquinase (galactokinase) resultando em danos principalmente nos olhos. A incidência é de 1: crianças, nas quais desde a infância podem se formar cataratas. Presença do gene autossômico recessivo na família é o principal fator de risco. Smith W. Pediatric Gastrointestinal Disease 2004; 55: , fourth edition
 resultando em danos principalmente nos olhos. A incidência é de 1: crianças, nas quais desde a infância podem se formar cataratas. Presença do gene autossômico recessivo na família é o principal fator de risco. Smith W. Pediatric Gastrointestinal Disease 2004; 55: , fourth edition.")
43
Características clínicas:
A patogênese é descrita pelo defeito da galactoquinase, o que gera acúmulo de galactose no sangue e nos tecidos. Características clínicas: catarata, anormalidades do sistema nervoso central: retardo mental, neurofibromatose, deterioração neurológica, epilepsia e pseudotumor cerebral. J Inherit Metab Dis;25(8):629-34,2002 Dec
:629-34,2002 Dec.")
44
C) Galactosemia tipo 3 É a deficiência do metabolismo da galactose caracterizado pela deficiência da atividade da uridil difosfo galactose-4-epimerase, resultando em 2 formas da doença, a saber: 1- forma inicial; 2- forma grave. A incidência é muito rara, tendo como fator de risco a presença do gene autossômico recessivo nos pais. Hum Mutat;23(4):396,2004 Apr
:396,2004 Apr.")
45
A patogênese é descrita como o defeito genético da uridil difosfo galactose-4-epimerase, o que gera acúmulo de galactose. As duas formas da doença são diferentes. Enquanto que na forma inicial os pacientes são assintomáticos (enzima deficiente apenas nas células do sangue, sendo normal nos outros tecidos), a forma grave apresenta pacientes com sintomas clínicos idênticos a forma da Galactosemia tipo 1. Mol Genet Metab;71(1-2):62-5,2000Sep
, a forma grave apresenta pacientes com sintomas clínicos idênticos a forma da Galactosemia tipo 1. Mol Genet Metab;71(1-2):62-5,2000Sep.")
46
Diagnóstico Galactosemia tipo 1: manifestações clínicas.
pesquisa de açúcar redutor na urina. ausência ou deficiência de GALT nos eritrócitos. no período pré natal – estudos enzimáticos em culturas de células obtidas por amniocentese ou pela comprovação de galactitol no líquido amniótico.

47
formação de catarata em lactentes e crianças.
Galactosemia tipo 2 formação de catarata em lactentes e crianças. substâncias redutoras diferentes da glicose na urina. deficiência de galactoquinase nos eritrócitos. Smith W. Pediatric Gastrointestinal Disease 2004; 55: , fourth edition

48
Tratamento Dieta isenta de galactose e lactose.
Monitoramento constante do nível de galactose e seus metabólitos no sangue do paciente, deve ser feito para verificação da eficácia da dieta. Monitoramento multidisciplinar. Smith W. Pediatric Gastrointestinal Disease 2004; 55: , fourth edition

49
III) Intolerância Hereditária à Frutose (Deficiência de frutose 1-fosfato Aldolase)
É um distúrbio autossômico recessivo, o qual é causado por mutações no gene humano Aldolase B, normalmente expressado no fígado, rins e intestino delgado. A mutação mais comum que acomete cerca de 53% das IHF dos alelos identificados ao redor do mundo, e resulta na substituição de Pro por Ala na posição 149. Mol Genet Metab;85(3):165-7,2005 Jul
:165-7,2005 Jul.")
50
A sua incidência é de 1: 20.000 nascidos vivos
O gene Aldolase B está localizado no braço longo do cromossomo 9(9q22.3). A sua incidência é de 1: nascidos vivos Esta enzima cataliza a conversão de frutose 1,6 fosfato e frutose 1- fosfato para triose fosfato. O defeito genético induz o acúmulo de frutose 1-fosfato (F-1-P), redução do fosfato intracelular e disponibilidade de ATP, resultando direto do seqüestro do fosfato como F-1-P. Mol Genet Metab;85(3):165-7,2005 Jul J Mol Biol;347(1):135-44,2005 Mar
. A sua incidência é de 1: nascidos vivos. Esta enzima cataliza a conversão de frutose 1,6 fosfato e frutose 1- fosfato para triose fosfato. O defeito genético induz o acúmulo de frutose 1-fosfato (F-1-P), redução do fosfato intracelular e disponibilidade de ATP, resultando direto do seqüestro do fosfato como F-1-P. Mol Genet Metab;85(3):165-7,2005 Jul. J Mol Biol;347(1):135-44,2005 Mar.")
51
Os altos níveis de F-1-P encontrados nos pacientes com deficiência de Aldolase inibem tanto a gliconeogênese como a glicogenólise, contribuindo para a hipoglicemia. O início da doença ocorre quando alimentos contendo sacarose/frutose são introduzidos na dieta. Aguda: lactentes desenvolvem irritabilidade, cólicas, vômitos, letargia, convulsões e coma.

52
Crônica: icterícia, edema, vômitos, diarréia, diminuição do apetite, hipodesenvolvimento, sepse, coagulopatia e hepatomegalia. Os achados laboratoriais, que são reversíveis com uma rigorosa exclusão da frutose da dieta, incluem: hipoglicemia, hipofosfatemia, aumento das aminotransferases séricas, hiperbilirrubinemia, hiperuricemia, aumento da excreção de uratos, acidose hiperclorêmica, hiperlactatemia, hipopotassemia, hipermagnesemia, anemia e trombocitopenia. J Inherit Metab Dis;25(7):571-5,2002 Nov Arch Biochem Biophys;408(2): ,2002 Dec
:571-5,2002 Nov. Arch Biochem Biophys;408(2): ,2002 Dec.")
53
Diagnóstico Requer a obtenção de uma história nutricional detalhada, correlacionando o início dos sintomas com a ingestão de alimentos contendo frutose. Presença de substâncias redutoras na urina. Medida direta da frutose 1- P- aldolase no tecido hepático. A histologia revela presença de esteatose associada a fibrose portal não inflamatória e acentuada formação pseudoacinar que ocasiona a ruptura dos cordões de hepatócitos.

54
Presença de cilindros biliares e fibrose portal e lobular
Presença de cilindros biliares e fibrose portal e lobular. Pode desenvolver cirrose. Aversão a alimentos doces é freqüente. Smith W. Pediatric Gastrointestinal Disease 2004; 55: , fourth edition Tratamento Consiste em eliminar todas as fontes de frutose da dieta, inclusive a sacarose e o sorbitol. Lentze MJ et al Am J Clin Nutr 1995;61:946-51

55
Conclusão A intolerância aos carboidratos é um problema cada vez mais freqüente e merece atenção especial dos gastroenterologistas pediátricos, pediatras e demais especialidades médicas, visando o diagnóstico e tratamento correto e precoce, minimizando, assim, as conseqüências da exposição desnecessária aos mesmos. Smith W. Pediatric Gastrointestinal Disease 2004; 55: , fourth edition

Apresentações semelhantes