Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Estabilidade de Formulações Farmacêuticas
Dr. UBIRACIR F. LIMA Vigilância Sanitária P&D Analítico prof.: Ubiracir F. Lima

2
Tecnologia Farmacêutica Eficácia terapêutica Segurança
Medicamento Pré Formulação: - Idealizar formulação - Identidade de ativo e excipientes - Funcionalidade - Características: moléculas; partículas; - Compatibilidade - Fornecedores Desintegrante Lubrificante Antioxidante… Ativo Aglutinante Processo Produtivo Tecnologia Farmacêutica Eficácia terapêutica Segurança Especificações Técnicas L M Cabral prof.: Ubiracir F. Lima

3
Estudo de Estabilidade
1. Objetivo O que é ? Estudo de Estabilidade Porque avaliar ? prof.: Ubiracir F. Lima

4
Perigo e Risco PRAZO DE VALIDADE DE FORMULAÇÕES Teor (%) Lote C Lote A
10% Eficácia Segurança Lote C Lote A Lote B 2 12 24 Tempo (meses) prof.: Ubiracir F. Lima
 prof.: Ubiracir F. Lima.")
5
O que é estabilidade ? Estabilidade farmacêutica Estabilidade química
resistência de uma dada formulação à alterações técnicas, durante o tempo em que se pretende comercializar o produto (dureza, dissolução, desagregação, viscosidade, e outros). resistência de ativos e excipientes a sofrer alterações em sua estrutura (reações), durante o tempo em que se pretende comercializar o produto. prof.: Ubiracir F. Lima
. resistência de ativos e excipientes a sofrer alterações em sua estrutura (reações), durante o tempo em que se pretende comercializar o produto. prof.: Ubiracir F. Lima.")
6
Porque avaliar ? Um medicamento é capaz de causar risco à saúde?
Parte da documentação necessária para registro e comercialização dos produtos (prazo de validade /AVISA) Suporte ao DT (validação das formulações desenvolvidas) Vigilância Sanitária – “conjunto de ações capazes de prevenir, reduzir ou eliminar riscos à saúde...” (Lei 8080/90) Um medicamento é capaz de causar risco à saúde? prof.: Ubiracir F. Lima
 Suporte ao DT (validação das formulações desenvolvidas) Vigilância Sanitária – conjunto de ações capazes de prevenir, reduzir ou eliminar riscos à saúde... (Lei 8080/90) Um medicamento é capaz de causar risco à saúde prof.: Ubiracir F. Lima.")
7
Encontro da Associação de Medicamentos Parenterais – Filadélfia, 1968.
“O Conceito de Data de Expiração de Medicamentos” Ernest G. Wollish, Bull Parenter Drug Assoc. 1969, 23(1), p Hoffman-La Roche, Nutley, NJ. prof.: Ubiracir F. Lima
, p Hoffman-La Roche, Nutley, NJ. prof.: Ubiracir F. Lima.")
8
Método Analítico Indicador
controle do tempo durante o qual um medicamento se mantém sem perder sua potência declarada em rótulo; Reação química Constituinte da embalagem x componente da formulação O2, CO2 Nenhuma decomposição deve resultar em redução da segurança Danos renais Tetraciclina anidrotetraciclina e epiteraciclina Frimpter, A.W., JAMA, 1963,184, 111-3 Método Analítico Indicador de Estabilidade ? prof.: Ubiracir F. Lima

9
Para uma empresa que tem como principal interesse garantir que seus produtos não estão alterados ou sendo vendidos em uma condição potencialmente adulterada, uma política conservadora , enfatizando principalmente segurança e estabilidade, aparece como sendo abordagem mais ética e prudente. prof.: Ubiracir F. Lima

10
2. Histórico Década de 50 – Período de intensas descobertas e lançamentos de novos medicamentos O uso da cinética química já parecia ser uma alternativa científica para a previsão do prazo de validade dos medicamentos. Por meio da ordem da reação de decomposição de substâncias químicas, em função da temperatura, pode-se calcular o tempo necessário para que ocorra uma redução de 10% do teor original. Este tempo é caracterizado como prazo de validade de um medicamento. Limitação – este tipo de determinação somente pode ser feita para substâncias puras e dissolvidas em água. (Moretto, L.D., Pharm. Technol., 3, 46-8, 1999) prof.: Ubiracir F. Lima
 prof.: Ubiracir F. Lima.")
11
Primeiros trabalhos T. Higushi, J. Am. Pharm. Assoc., 12, 707, 1953 –
Determinação da cinética de degradação da solução do antibiótico Cloranfenicol, observando perda da atividade. Neste trabalho foi monitorada a velocidade de formação do íon cloreto (Cl-) em meio aquoso. prof.: Ubiracir F. Lima
 em meio aquoso. prof.: Ubiracir F. Lima.")
12
Estabilidade histórico
E.R. Garret, J. Am. Pharm. Assoc., 12, 684, 1959 – Estudo dos produtos, e mecanismos, da degradação pirolítica do Anidrido de Aspirina. -Estudos em 40 oC, 50 oC, 60 oC, 70 oC e 4oC -Material de embalagem - contato com a substância ativa -Umidade residual -IV – 1700 – 1800 cm-1 ( região de carbonila) prof.: Ubiracir F. Lima
 prof.: Ubiracir F. Lima.")
13
. Até 1984 metodologias próprias, reunião de dados e
informações na documentação de registro princípios técnicos e científicos sem interferências de atos regulatórios Processo de especialização das unidades produtivas Comércio internacional Globalização Racionalização da produção Reconhecimento das zonas climáticas Estímulo nas autoridades da saúde e entidades do setor industrial farmacêutico, a buscar uma racionalização na coleta de dados para compor os documentos necessários para registro, dentre os quais o da estabilidade. prof.: Ubiracir F. Lima

14
. Década de 80 – surgimento de alguns regulamentos utilizados para a
previsão do prazo de validade de fármacos e medicamentos. . ampliar o conhecimento sobre a complexidade do assunto Multiplicidade de documentos confusão nos profissionais envolvidos: atividades técnicas, preparação de documentos encarregados na avaliação de documentação de registro prof.: Ubiracir F. Lima

15
Singh, S., Pharma. Technol., 68-88, 1999
PAÍS GUIDELINE ANO DE INTRODUÇÃO Japão Standards for Stability Testing of New Drugs (1984) UK Guidance Notes on Applications for Products Licenses USA Submitting Documentation for the Stability of Human Drugs and Biologicals UE Stability Testing on Active Ingredients and Finished Products Singh, S., Pharma. Technol., 68-88, 1999 prof.: Ubiracir F. Lima
 UK Guidance Notes on Applications for Products Licenses USA Submitting Documentation for the Stability of Human. Drugs and Biologicals UE Stability Testing on Active Ingredients and Finished Products Singh, S., Pharma. Technol., 68-88, prof.: Ubiracir F. Lima.")
16
. Década de 90 1991 – New Trends and Requirements – An International
Symposium for the EU, USA and Japan. 2005 – ANVISA – RE 01 2004 – ANVISA – RE 398 1991 – First International Conference on Harmonization (ICH1) Stability Testing of New Drug Substances and Products – ICH (R2) Harmonização Japão, USA e EU. “Brasil” 1993 – Stability Testing of New Drug Substances and Products – (ICH2) 2002 – ANVISA – RE 560 Resolução GMC Nº 53/96 Resolução 391/99 prof.: Ubiracir F. Lima
 Stability Testing of. New Drug Substances. and Products – ICH (R2) Harmonização. Japão, USA e EU. Brasil 1993 – Stability Testing of. New Drug Substances and. Products – (ICH2) 2002 – ANVISA – RE Resolução GMC Nº 53/ Resolução 391/99. prof.: Ubiracir F. Lima.")
17
. RDC nº 210, de 04 de agosto de 2003 – REGULAMENTO TÉCNICO DAS BOAS PRÁTICAS PARA A FABRICAÇÃO DE MEDICAMENTOS 1.3 O fabricante é responsável pela qualidade dos medicamentos por ele fabricados, assegurando que estes são adequados aos fins aos quais se destinam, cumprem com os requisitos estabelecidos em seu registro e não colocam os pacientes em risco por apresentar segurança, qualidade ou eficácia inadequadas.... Todo o pessoal deve conhecer os princípios das BPF e receber treinamento inicial e contínuo... Todo o pessoal deve ser motivado a apoiar a empresa na manutenção dos padrões de qualidade. prof.: Ubiracir F. Lima

18
16.6 Estudo de estabilidade
O Controle de qualidade deve avaliar a qualidade e a estabilidade dos produtos terminados e, quando necessário, das matérias-primas, dos produtos intermediários e a granel. O Controle de qualidade deve fixar as datas de vencimento e as especificações quanto ao prazo de validade, tendo como base os ensaios de estabilidade realizados de acordo com as condições de armazenamento (RE 398). prof.: Ubiracir F. Lima
. prof.: Ubiracir F. Lima.")
19
. 16.6.3 Deve ser desenvolvido e Implementado um programa
(a) descrição completa do produto envolvido no estudo; . b) todos os parâmetros dos métodos e dos ensaios, que devem descrever os procedimentos dos ensaios de potência, de pureza e as características físicas, bem como as evidências documentadas de que os ensaios realizados são indicadores da estabilidade do produto; (c) previsão quanto a inclusão de um número suficiente de lotes; Deve ser desenvolvido e Implementado um programa escrito de estudo de estabilidade, incluindo os seguintes elementos: (d) cronograma de ensaio para cada produto; (e) instruções sobre condições especiais de armazenamento; (f) instruções quanto à retenção adequada de amostras; e (g) um resumo de todos os dados obtidos, incluindo a avaliação e as conclusões do estudo. A estabilidade de um produto deve ser determinada antes da comercialização e deve ser repetidos após quaisquer mudanças significativas nos processos de produção, equipamentos, materiais de embalagem, etc. prof.: Ubiracir F. Lima
 descrição completa do produto envolvido no estudo; . b) todos os parâmetros dos métodos e dos ensaios, que devem. descrever os procedimentos dos ensaios de potência, de pureza. e as características físicas, bem como as evidências documentadas. de que os ensaios realizados são indicadores da estabilidade. do produto; (c) previsão quanto a inclusão de um número. suficiente de lotes; Deve ser desenvolvido e. Implementado um programa. escrito de estudo de estabilidade, incluindo os seguintes elementos: (d) cronograma de ensaio para. cada produto; (e) instruções sobre condições. especiais de armazenamento; (f) instruções quanto à retenção. adequada de amostras; e. (g) um resumo de todos os dados obtidos, incluindo a avaliação e as conclusões do estudo A estabilidade de um produto deve ser determinada antes da. comercialização e deve ser repetidos após quaisquer mudanças significativas. nos processos de produção, equipamentos, materiais de embalagem, etc. prof.: Ubiracir F. Lima.")
20
. GMP – Gerenciamento administrativo Amostras Documentação
quantidade suficiente para cobrir todos os testes sistema de alerta para data de testes armazenamento de resultados analíticos e históricos Espaço físico nas câmaras climáticas prof.: Ubiracir F. Lima

21
Amostras prof.: Ubiracir F. Lima

22
. GMP - Gerenciamento Técnico Equipamentos Analistas Treinamento /
capacitação Volume efetivo dimensionado Método analítico (Cromatografia) Exclusividade / histórico Qualificação Validação prof.: Ubiracir F. Lima
 Exclusividade / histórico. Qualificação. Validação. prof.: Ubiracir F. Lima.")
23
Considerações Gerais Reconhecimento das características da substância ativa. Reatividade dos grupamentos funcionais Elucidação dos produtos de decomposição (submeter amostras a condições favoráveis a formação) Avaliação toxicológica – Determinação de limites de especificação Desenvolvimento de metodologias analíticas Métodos cromatográficos Validação das metodologias eleitas prof.: Ubiracir F. Lima
 Avaliação toxicológica – Determinação de limites de especificação. Desenvolvimento de metodologias analíticas. Métodos cromatográficos. Validação das metodologias eleitas. prof.: Ubiracir F. Lima.")
24
Pré-requisitos para realização de testes de Estabilidade.
Produtos em lançamento – ANVISA Alteração na formulação Troca de fornecedores/fabricantes de matérias primas, especialmente do princípio ativo Alterações ou substituições de equipamentos que afetem a qualidade do produto prof.: Ubiracir F. Lima

25
Trocas ou modificações nas instalações físicas e nos sistemas de apoio, crítico da área da produção
Mudanças de tamanho de lotes; Trocas ou modificações dos procedimentos de manutenção e de limpeza dos equipamentos e da área de fabricação comprometidas com o produto; Modificações ou troca da embalagem primária e fechamento Troca de fornecedor de materiais da embalagem primária e fechamento sempre que modifiquem as características do material

26
Uso de lote comparativo
Reprocesso de produtos De forma periódica programada, como uma política de checagem da fabricação de um produto; ou os 3 primeiros lotes de produção de um produto novo ou de um processo padronizado e validado; Quando houver alterações relevantes no processo de fabricação. Follow-Up – FDA Uso de lote comparativo

27
Vivência no laboratório:
. Estudo Racional da Estabilidade dos Fármacos Prático Científico Vivência no laboratório: DT + CQ Conhecimento das Propriedades dos insumos Adaptação de relatos da literatura Domínio da metodologia analítica empregada Processo ágil e eficaz prof.: Ubiracir F. Lima

28
Estabilidade e as Propriedades Farmacêuticas
O termo “estabilidade farmacêutica”, engloba vários conceitos. Foi primeiro aplicado à estabilidade química de um fármaco em uma formulação adequada. Entretanto a performance de um fármaco, quando formulado como um comprimido, cápsula, solução, suspensão, não depende unicamente do conteúdo do fármaco, mas também, de suas propriedades farmacêuticas (dureza, tempo de dissol., desag.). Todos estes aspectos devem ser parte de um programa de estabilidade. prof.: Ubiracir F. Lima
. Todos estes aspectos devem ser parte de um programa de estabilidade. prof.: Ubiracir F. Lima.")
29
. Dissolução X Biodisponibilidade Eficácia terapêutica Sólido
Parenteral ! Testes Físicos Solução Material precipitado Queda no teor ? Sol. oral Mercado prof.: Ubiracir F. Lima

30
a) Soluções Importantes fatores:
Aparência Importantes fatores: Propriedades organolépticas Para avaliação organoléptica é importante estabelecer um painel teste Treinar mais que um analista para realização destes testes capacidade orgânica (análise sensorial); soluções diluídas de quinina Odor diferenças por pequenas sutilezas
; soluções diluídas. de quinina. Odor. diferenças por pequenas sutilezas.")
31
Sabor – Aparência – Descrição: Grau de acidez Grau de salinidade
um bom analista sensorial consegue duplicar seus resultados e lembrar-se de um sabor avaliado a 3 meses. Descrição: Grau de acidez Grau de salinidade Nível de sabor Tipo de sabor Aparência – soluções, podem ter uma tendência à alteração na cor. Muitas vezes não é possível, com sensibilidade analítica, estabelecer a fonte da coloração, nem o nível de substância que a causa. Cromóforos / estado de oxidação de metais padrões de cores ! prof.: Ubiracir F. Lima

32
Muitos processos de descoloração são de natureza oxidativa e catalisadas por íons metálicos.
uso de antioxidantes. Acetilcisteína 0,5% Ác. Ascórbico 0,02 – 1% Ác. Cítrico (quelante) composição variável EDTA-Na 0,01 – 0,75% Mendenhall (Drug. Dev. Ind. Pharm., 1984, 10, 1297) prof.: Ubiracir F. Lima
 composição variável. EDTA-Na 0,01 – 0,75% Mendenhall (Drug. Dev. Ind. Pharm., 1984, 10, 1297) prof.: Ubiracir F. Lima.")
33
Em regra geral: suspensões vão separar.
b) suspensões Em regra geral: suspensões vão separar. Velocidade de sedimentação Volume de sedimentação A composição da embalagem pode ser fator crítico na estabilidade das suspensões. prof.: Ubiracir F. Lima
 suspensões. Em regra geral: suspensões vão separar. Velocidade de sedimentação. Volume de sedimentação. A composição da embalagem pode ser fator crítico na estabilidade das suspensões. prof.: Ubiracir F. Lima.")
34
C) Sistemas semi-sólidos (pomadas, cremes e supositórios)
Fatores para checar em um programa de estabilidade de sistemas semi-sólidos: Consistência Viscosidade Polimorfismo Davis (Stability Testing of Drug Products ) prof.: Ubiracir F. Lima
 prof.: Ubiracir F. Lima.")
35
d) Comprimidos Propriedades físicas associadas:
dureza, desintegração, dissolução e aparência Dureza freqüentemente comprimidos se tornarão tanto moles quanto duros em pequenos espaços de tempo após a produção. O amolecimento provém da expansão das “ligações” cruzadas adquiridas na etapa de compressão O aumento na dureza é decorrente da cristalização de compostos solúveis em pequena quantidades de umidade absorvidas. prof.: Ubiracir F. Lima

36
Desintegração e dissolução
Se um comprimido desintegra por virtude de um agente desintegrante que expande quando umidecido, então o fator mais importante é a velocidade com que o líquido desintegrante penetra no comprimido, o desintegra e permite que os ativos sejam solubilizados. (Covreur, tese de doutoramento na Univ. Catholique de Louvrain – Belgica) Etapa de adição do agente desintegrante. prof.: Ubiracir F. Lima
 Etapa de adição do agente desintegrante. prof.: Ubiracir F. Lima.")
37
Trata-se de estabilidade a ser monitorada em função do tempo
Aparência Trata-se de estabilidade a ser monitorada em função do tempo Muitas vezes é observado com descrição subjetiva. métodos quantitativos Ficha ou quadro de cores (Rothgang, 1974, Dtsc. Apoth. Ztg.) Avaliação espectrofotmetrica ( Hammouda, 1971, Pharmazie) Fotografia (Armstrong, 1974, J. Pharm. Sci.) Alteração na coloração Aparecimento de pontos escuros prof.: Ubiracir F. Lima
 Avaliação espectrofotmetrica ( Hammouda, 1971, Pharmazie) Fotografia (Armstrong, 1974, J. Pharm. Sci.) Alteração na coloração. Aparecimento de pontos escuros. prof.: Ubiracir F. Lima.")
38
Estabilidade e as Propriedades Químicas - Estruturas dos Fármacos
Estabilidade - Resistência a reações químicas, principalmente de ocorrência nos constituintes ativos das formulações. Objetivo: determinação de prazo de validade! Tempo em que se observa uma redução de 10% na potência declarada em rótulo. prof.: Ubiracir F. Lima

39
Moretto ( Pharm. Technol.- 1999)
O detentor do registro do medicamento é responsável pela manutenção das características especificadas perante o órgão de vigilância sanitária e perante a sociedade. ü De acordo com os conceitos internacionais, um medicamento deve ser eficaz e seguro, e ter as características de qualidade conforme definido no registro. prof.: Ubiracir F. Lima

40
“Na natureza nada se perde e nada se cria. Tudo se transforma”
Antoine Lavoisier ( ) - lei da conservação das massas prof.: Ubiracir F. Lima
 - lei da conservação das massas. prof.: Ubiracir F. Lima.")
41
. Qualquer alteração na estrutura do fármaco pode reduzir sensivelmente a atividade terapêutica, bem como levar a efeitos indesejados. vitamina C 1g mg mg prof.: Ubiracir F. Lima

42
. Ferramentas Porque as substâncias se transformam em outras ?
Como se processa essa transformação ? Ocorre o rompimento de algumas ligações e formação de novas ! Fatores que afetam a estabilidade das formulações Processo de degradação Estabilidade química prof.: Ubiracir F. Lima

43
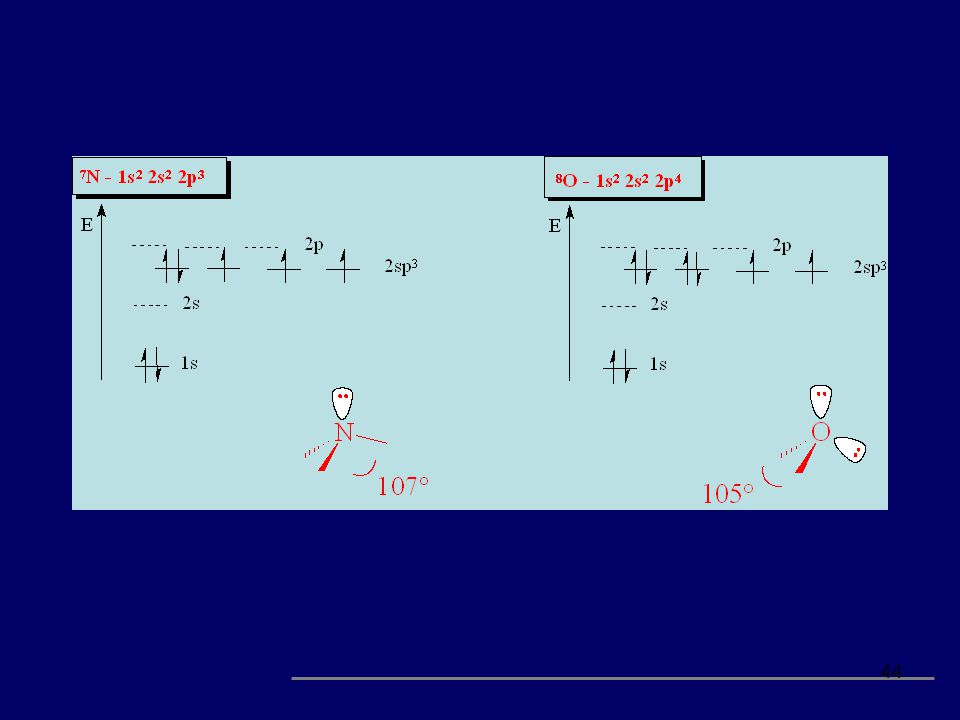
4.1 - Química Orgânica – Fundamentos
Parte da química que estuda os componentes de carbono. Estrutura atômica C, H, N, O prof.: Ubiracir F. Lima

45
Estrutura molecular – formação e quebra das ligações
a) Formação de ligações (pq se formam?) Níveis de Energia Preenchimento orbitais prof.: Ubiracir F. Lima
 Formação de ligações (pq se formam ) Níveis de Energia. Preenchimento orbitais. prof.: Ubiracir F. Lima.")
46
CH4 . prof.: Ubiracir F. Lima

47
prof.: Ubiracir F. Lima

48
prof.: Ubiracir F. Lima

49
b) Rompimento de ligações (pq se rompem?)
Lig. π p-p = 63 Kcal/mol Lig. σ sp2-sp2 = 83 Kcal/mol H2C=CH2 A ligação π é 13% mais fraca que a lig. Um alceno será mais reativo que um alcano ! prof.: Ubiracir F. Lima

50
. H2C=O Lig. π p-p = 82 Kcal/mol Lig. σ sp2-sp2 = 91 Kcal/mol
Nesta carbonila, a ligação π é 9% mais fraca que a lig. A carbonila será mais reativa frente a um álcool ! prof.: Ubiracir F. Lima

51
Estabilidade química Polaridade da Ligação
Ligação covalente - há compartilhamento de elétrons Eletronegatividade - capacidade de um átomo atrair elétrons H = 2,1 C = 2, N = 3, O = 3,5 prof.: Ubiracir F. Lima

52
Estabilidade química Exemplos: Apolar Polar
Esta polarização das ligações resulta em deficiência eletrônica no carbono da carbonila, conferindo maior reatividade a esta função! prof.: Ubiracir F. Lima

53
Estabilidade química prof.: Ubiracir F. Lima

54
4.2- Funções orgânicas sensíveis à degradação
álcool éter aldeído prof.: Ubiracir F. Lima

55
éster lactona amida H - α prof.: Ubiracir F. Lima

56
Bleomicina (carciomas)
prof.: Ubiracir F. Lima

57
5. Reações que causam degradação
Variedade de compostos utilizados como fármacos diversidade estrutural Grupamentos funcionais Diversidade nas espécies de reações de decomposição são possíveis. Para antecipar os problemas de estabilidade, deve-se ser capaz de identificar as potenciais reações e estimar a reatividade dos fármacos prof.: Ubiracir F. Lima

58
Apesar de um grande número de reações levarem à degradação, a sua maioria pode ser classificada como reações de HIDRÓLISE e OXIDAÇÃO. grupos funcionais comuns nos fármacos ocorrência natural de umidade e oxigênio prof.: Ubiracir F. Lima

59
5.1 – Hidrólise São reações que envolvem adição de uma molécula de água resultando em clivagem do fármaco. Exemplos farmacêuticos de substâncias que degradam por hidrólise incluem ésteres como a Aspirina, amidas como o Cloranfenicol, lactamas como as Penicililnas e outros. prof.: Ubiracir F. Lima

60
prof.: Ubiracir F. Lima

61
prof.: Ubiracir F. Lima

62
5.2 – Oxidação O termo oxidação é aplicado a reações em que um ou mais átomos eletropositivos, radicais ou elétrons são perdidos, ou quando um ou mais átomos eletronegativos ou radicais são ganhos. prof.: Ubiracir F. Lima

63
Reação redox Transferência reversível de elétrons
sem adição ou remoção de O2. Em certas formulações, pode-se prever a reatividade de uma substância frente a este processo de oxidação através de seu potencial padrão de oxidação εo. prof.: Ubiracir F. Lima

64
Em solução aquosa de ác. Ascórbico (ε0 = +0,14v) e adrenalina (ε0 = +0,52v), ocorre oxidação do ácido. prof.: Ubiracir F. Lima

65
auto-oxidação Trata-se de uma reação irreversível em que uma substância torna-se oxidada na presença de oxigênio atmosférico. Radicais livres Na fabricação e estocagem de produtos farmacêuticos, auto-oxidações são mais comuns que reações redox. prof.: Ubiracir F. Lima

66
prof.: Ubiracir F. Lima

67
5.3 – Isomerização geométrica e óptica
Neste processo de isomerização há conversão de uma substância no seu isômero. Isômero – são substâncias que possuem a mesma fórmula molecular, porém diferem estruturalmente ESTEREOQUÍMICA prof.: Ubiracir F. Lima

68
Isomeria geométrica Esta conversão envolve troca na configuração relativa espacial de átomos ou grupos ligados a duplas ligações etilênicas, ou a compostos cíclicos. Diferentes isômeros podem possuir diferentes potências ou até diferentes propriedades no organismo ! prof.: Ubiracir F. Lima

69
A forma mais ativa da Vitamina A possui configuração trans
A forma mais ativa da Vitamina A possui configuração trans. Em solução aquosa, isomeriza para formar os isômeros menos ativos 6-cis e 2,6-di-cis. prof.: Ubiracir F. Lima

70
Cerca de 40% das drogas comercializadas são quirais !
Isomeria óptica Envolve a troca na configuração relativa de átomos ou grupos em torno de um carbono assimétrico. Cerca de 40% das drogas comercializadas são quirais ! (Ferreira, V.F. e Pinheiro, S.; Química Nova, 1999) prof.: Ubiracir F. Lima
 prof.: Ubiracir F. Lima.")
71
Carbono assimétrico (*) Contém 4 substituintes diferentes
Possuem imagem especular não sobreponível prof.: Ubiracir F. Lima

72
Fenômeno óptico – estes estereoisômeros são capazes de girar plano da luz plano-polarizada Cada enanciômero é capaz de desviar o plano da luz polarizada em igual amplitude, porém em sentido oposto. prof.: Ubiracir F. Lima

73
Dois tipos de isomerismo óptico são distinguidos em produtos farmacêuticos:
RACEMIZAÇÃO EPIMERIZAÇÃO Racemização – envolve a conversão de um fármaco opticamente ativo em seu isômero. A reação de conversão prossegue até a concentração dos isômeros se igualarem atingindo o equilíbrio – [α ] = zero prof.: Ubiracir F. Lima

74
A atividade farmacológica do isômero levorotatório (-) da adrenalina em sol. Aquosa, é cerca de 15 vezes maior que a forma destro. prof.: Ubiracir F. Lima

75
Nem sempre o processo de isomerização conduz
unicamente à redução da atividade farmacológica. Os receptores biológicos irão reconhecer de forma seletiva os diferentes estereoisômeros. prof.: Ubiracir F. Lima

76
A talidomida nos anos 50 causou sérios problemas quando administrado em sua forma isomérica errada.
S-talidomida (teratogênico) N O H HB R 2 B R-talidomida (ativo) prof.: Ubiracir F. Lima
 N. O. H. HB. R. 2. B. R-talidomida (ativo) prof.: Ubiracir F. Lima.")
77
Epimerização – racemização se processa em apenas um deles
mais de um centro quiral no fármaco o equilíbrio entre os isômeros não representa concentrações iguais favorececimento de um dos epímeros [α ] zero prof.: Ubiracir F. Lima

78
A tetraciclina epimeriza em solução ácida para formar 4-epitetraciclina, que possui atividade antimicrobiana reduzida. prof.: Ubiracir F. Lima

79
5.4 – Reações fotoquímicas
Fármacos Luz Reações Fotoquímicas Excipientes Isomerização Ciclização Polimerizaçao prof.: Ubiracir F. Lima

80
Isomerização promoção de elétrons π até orbitais
moleculares antiligantes (ψ*) Isomerização rotação em torno da ligação σ Por exemplo, na isomerização do antifúngico ac. trans-cinâmico. prof.: Ubiracir F. Lima
 Isomerização. rotação em torno da ligação σ. Por exemplo, na isomerização do antifúngico ac. trans-cinâmico. prof.: Ubiracir F. Lima.")
81
prof.: Ubiracir F. Lima

82
Ciclização – uma nova ligação será formada com um carbono
Ciclização – uma nova ligação será formada com um carbono de um centro vizinho. O ácido meclofenâmico sofre desidrohalogenação, e cicliza levando a formação de dois novos produtos. prof.: Ubiracir F. Lima

83
Polimerização – A combinação de duas ou mais moléculas idênticas para formar uma molécula mais complexa é conhecida como polimerização. Nos produtos farmacêuticos, este processo é responsável, além da perda de atividade farmacológica, é também responsável pelo surgimento de precipitados insolúveis nas soluções. prof.: Ubiracir F. Lima

84
A estocagem a frio de uma solução aquosa de formaldeído pode
resultar no aparecimento de um depósito branco, que consiste no paraformaldeído formado por polimerização. prof.: Ubiracir F. Lima

85
5.5 - Interação Fármaco-Excipiente e Fármaco-Fármaco
quase que exclusivamente em fase líquida na superfície do cristal processo de degradação catalisador de superfície excipientes Ação Alterando o pH da fase líquida Reagindo diretamente com o fármaco prof.: Ubiracir F. Lima

86
Reação entre dois fármacos
A maioria das ocorrências envolvem trans-acilação de um fármaco, com outro contendo grupamentos hidroxila ou amina. prof.: Ubiracir F. Lima

87
Comprimidos em camadas
Na prática, este problema de reação entre um doador–acil ativo e um aceptor, pode ser prevenido por separação física das espécies reativas: Comprimidos em camadas prof.: Ubiracir F. Lima

88
6. Fatores que afetam a estabilidade química e métodos de estabilização
Para se obter um aumento da estabilidade de um fármaco, é necessário um conhecimento seguro da natureza das reações de degradação passíveis de ocorrência durante a produção e estocagem, e dos fatores que podem acelerar a velocidade da degradação. Uma vez que muitos fármacos são complexos em termos físico-químicos, um vasto trabalho experimental é necessário para alcançar uma estabilidade química aceitável, e então prever o prazo de validade dos produtos. prof.: Ubiracir F. Lima

89
Estágios importantes para monitorar condições de estocagem
Pré-formulação Aplicação do CQ excipientes Estágios importantes para monitorar Desenvolvimento do processo Avaliação de embalagem prof.: Ubiracir F. Lima

90
Fatores que afetam a velocidade de reação química
De um Fármaco ou Excipientes pH Natureza do Solvente Concentração Produção transporte Temperatura Estocagem prof.: Ubiracir F. Lima

91
pH – Conhecer os efeitos do pH na velocidade de degradação
permite ao formulador ajustar o pH próximo àquele correspondente ao máximo de estabilidade. A oxidação da adrenalina injetável é minimizada pelo uso de ácido tartárico (pH 2,8-3,6) próximo ao pH de máxima estabilidade (pH 3,2-3,4) prof.: Ubiracir F. Lima
 próximo ao pH de máxima estabilidade (pH 3,2-3,4) prof.: Ubiracir F. Lima.")
92
Efeitos irritantes também devem ser considerados.
A formulação de uma solução próximo ao pH de estabilidade máxima nem sempre é possível, devido à solubilidade (absorção por tecidos) e eficácia. Efeitos irritantes também devem ser considerados. Bases fracas de alcalóides como a homatropina, são mais estáveis em solução ácida (pH 3-4), porém apresentam melhores resultados de absorção e minimização de efeitos irritantes em solução levemente alcalina (pH 7,4). prof.: Ubiracir F. Lima
 e eficácia. Efeitos irritantes também devem ser considerados. Bases fracas de alcalóides como a homatropina, são mais estáveis em solução ácida (pH 3-4), porém apresentam melhores resultados de absorção e minimização de efeitos irritantes em solução levemente alcalina (pH 7,4). prof.: Ubiracir F. Lima.")
93
Em situações semelhantes, é possível selecionar um pH
intermediário (pH 5) com o intuito de alcançar estabilidade adequada, eficácia e conforto para o paciente. prof.: Ubiracir F. Lima
 com o intuito. de alcançar estabilidade adequada, eficácia e conforto para o paciente. prof.: Ubiracir F. Lima.")
94
Natureza do solvente – Dois fatores podem ser responsáveis pela estabilidade dos fármacos nestas situações: O deslocamento do equilíbrio favorecendo a formação dos produtos de degradação (polaridade dos solventes) Reatividade dos solventes frente aos fármacos. prof.: Ubiracir F. Lima
 Reatividade dos solventes frente aos fármacos. prof.: Ubiracir F. Lima.")
95
A polaridade de um solvente é responsável pela habilidade de solvatar espécies semelhantes.
De acordo com a teoria do estado de transição, o equilíbrio será deslocado na direção mais favorável energeticamente. prof.: Ubiracir F. Lima

96
experiência acumulada
solvente adequado adaptação à sistemas semelhantes literatura A solução da fenobarbitona de sódio em uma solução de propilenoglicol e água (9:1 v/v) pode ser esterilizada em autoclave, enquanto uma solução aquosa degrada apreciavelmente. prof.: Ubiracir F. Lima
 pode ser esterilizada em autoclave, enquanto uma solução aquosa degrada apreciavelmente. prof.: Ubiracir F. Lima.")
97
Interação Fármaco x Solvente
A tintura de arnica tem como principais responsáveis pela atividade anti reumática, lactonas sesquiterpênicas como a diidrohelenalina. Estes fitoterápicos em contato com o solvente etanol, sofrem adição de Michael tipo 1,4, afetando a eficácia farmacológica da tintura. Schmidt, T.J., Planta Med., 2000, 66, prof.: Ubiracir F. Lima

98
Concentração do fármaco –
A concentração do fármaco nas formulações usualmente é determinada por considerações terapêuticas, porém algumas vezes possibilita elevar a estabilidade pela sua modificação. Em suspensões de aspirina, onde a velocidade de degradação independe da concentração, o prazo de validade pode ser prolongado pelo incremento da concentração do fármaco, embora um concomitante aumento na viscosidade restringe a aplicação desta abordagem. prof.: Ubiracir F. Lima

99
Para a ampicilina, soluções diluídas são mais estáveis, uma vez que soluções concentradas favorecem o processo de dimerização. prof.: Ubiracir F. Lima

100
Controle da temperatura
O controle preciso da temperatura e o tempo de aquecimento no processo de esterilização, são especialmente importantes em produtos termo-lábeis. Para certos fármacos como a procaína, onde a energia de ativação do processo de degradação é significantemente menor que para a eliminação da maioria de bactérias, é preferível esterilizar as soluções por um período muito curto de tempo à temperatura elevada. prof.: Ubiracir F. Lima

101
Controle do conteúdo de oxigênio
Manter criteriosamente fechado as embalagens contendo o fármaco. Deslocar a atmosfera residual em frascos ou ampolas, por meio de passagem de gás inerte. Geralmente N2 prof.: Ubiracir F. Lima

102
Controle do conteúdo de água
Fármacos Instáveis em solução Reconstituição extemporânea liofilização secagem na compressão Processos adequados laqueamento microencapsulação com materiais hidrofóbicos monitoramento do teor de umidade nos excipientes. Materiais de embalagens impermeáveis Adição de sílica gel prof.: Ubiracir F. Lima

103
Anti-oxidantes São substâncias que adicionadas em pequena quantidade podem inibir o processo de oxidação Anti-oxidantes verdadeiros bloqueadores de reações radicalares Agentes redutores possuem menor potencial padrão de oxidação εo Anti-oxidantes sinérgicos atuam elevando o potencial de outros anti-oxidantes prof.: Ubiracir F. Lima

104
7 - Metodologia Analítica
Resolução nº 391, de 09 de agosto de 1999 RDC nº 10, de 02 de janeiro de 2001 GUIA PARA VALIDAÇÃO DE MÉTODOS ANALÍTICOS RDC nº 84, de 19 de março de 2002 RDC nº 135, de 29 de maio de 2003 RDC nº 210 de 04 de agosto de 2003 RE nº 475, de 19 de março de 2002 RE nº 899, de 29 de maio de 2003 prof.: Ubiracir F. Lima

105
2.1. Especificidade e Seletividade:
É a capacidade que o método possui de medir exatamente um composto em presença de outros componentes tais como impurezas, produtos de degradação e componentes da matriz. Para análise qualitativa (teste de identificação) é necessário demonstrar a capacidade de seleção do método entre compostos com estruturas relacionadas que podem estar presentes. Isto deve ser confirmado pela obtenção de resultados positivos (preferivelmente em relação ao material de referência conhecido) em amostras contendo o fármaco, comparativamente com resultados negativos obtidos com Amostras que não contém o fármaco, mas compostos estruturalmente semelhantes. prof.: Ubiracir F. Lima
 é necessário. demonstrar a capacidade de seleção do método entre compostos com. estruturas relacionadas que podem estar presentes. Isto deve ser. confirmado pela obtenção de resultados positivos (preferivelmente em. relação ao material de referência conhecido) em amostras contendo. o fármaco, comparativamente com resultados negativos obtidos com. Amostras que não contém o fármaco, mas compostos estruturalmente. semelhantes. prof.: Ubiracir F. Lima.")
106
Para análise quantitativa (teor) e análise de impurezas, a especificidade pode ser determinada pela comparação dos resultados obtidos de amostras (fármaco ou medicamento) contaminadas com quantidades apropriadas de impurezas ou excipientes e amostras não contaminadas, para demonstrar que o resultado do teste não é afetado por esses materiais. prof.: Ubiracir F. Lima

107
Quando a impureza ou o padrão do produto de degradação não estiverem disponíveis, pode-se comparar os resultados do teste das amostras contendo impurezas ou produtos de degradação com os resultados de um segundo procedimento bem caracterizado (por exemplo metodologia farmacopéica ou outro procedimento validado). Estas comparações devem incluir amostras armazenadas sob condições de estresse (por ex. luz, calor umidade, hidrólise ácida/básica, oxidação). prof.: Ubiracir F. Lima

108
Em métodos cromatográficos, deve-se tomar as precauções necessárias para garantir a pureza dos picos cromatográficos. A utilização de testes de pureza de pico (por exemplo, com auxilio de detector de arranjo de fotodiodos ou espectrometria de massas) são interessantes para demonstrar que o pico cromatográfico é atribuído a um só componente. prof.: Ubiracir F. Lima

109
8. Métodos de Ensaio Indicadores de Estabilidade
Stability-indicating assay methods (SIAM) Embora a exigência destes métodos estejam presentes em documentos regulatórios (RE 01/05), nem Farmacopéias ou Guias de agências reguladoras disponibilizam etapas básicas para serem seguidas no seu desenvolvimento e validação. HPLC 85-90% dos métodos citados na literatura prof.: Ubiracir F. Lima
 Embora a exigência destes métodos estejam presentes em documentos. regulatórios (RE 01/05), nem Farmacopéias ou Guias de agências reguladoras. disponibilizam etapas básicas para serem seguidas no seu. desenvolvimento e validação. HPLC % dos métodos citados na literatura. prof.: Ubiracir F. Lima.")
110
8.1 Métodos Cromatográficos –
Separação Identificação Quantificação prof.: Ubiracir F. Lima

111
Quantificar os produtos formados
prof.: Ubiracir F. Lima

112
HPLC prof.: Ubiracir F. Lima

113
Características de detecção - UV
prof.: Ubiracir F. Lima

114
A absorção da luz para soluções da mesma amostra é tanto maior quanto mais concentrada for a solução por ela atravessada: solução 10 g/l Io IT1 solução 20 g/l IT2 feixe de luz de intensidade Io A solução 20g/l absorve o dobro da solução 10g/l. prof.: Ubiracir F. Lima

115
Devido às suas estruturas moleculares, amostras diferentes absorvem diferentes quantidades de um mesmo tipo de luz ! prof.: Ubiracir F. Lima

116
ε = coeficiente de absorção molar
Diferentes tipos de luzes (λ) serão absorvidas de forma diferente por uma mesma amostra ! A m o s t r a ε = coeficiente de absorção molar prof.: Ubiracir F. Lima
 serão absorvidas de. forma diferente por uma mesma amostra ! A m o s t r a. ε = coeficiente de absorção molar. prof.: Ubiracir F. Lima.")
117
Devido às suas estruturas moleculares, amostras diferentes
absorvem diferentes quantidades de um mesmo tipo de luz ! prof.: Ubiracir F. Lima

118
prof.: Ubiracir F. Lima

119
Qualquer sinal presente no cromatograma deve ser identificado e a sua presença claramente
justificada ! prof.: Ubiracir F. Lima

120
Cromatografia em Camada Fina –
prof.: Ubiracir F. Lima

121
prof.: Ubiracir F. Lima

122
8.2 Etapa 1: estudo crítico da estrutura do fármaco para
prever processo da decomposição. Primeira etapa sempre que se inicia um projeto de desenvolvimento de um SIAM. Grupamento Funcional amidas ésteres hidrólise Categorias olefinas decomposição Foto lactonas Aril halo derivados lactamas álcool amina oxidação prof.: Ubiracir F. Lima

123
Estrutura de moléculas já conhecidas (bioisósteros)
novos fármacos Estrutura de moléculas já conhecidas (bioisósteros) Anel β-lactâmico n > 40 penicilinas prof.: Ubiracir F. Lima
 Anel β-lactâmico. n > 40 penicilinas. prof.: Ubiracir F. Lima.")
124
Hidrólise dos Bloqueadores α-adrenérgicos de estrutura similar
prof.: Ubiracir F. Lima

125
Atenção ! Alguns derivados sofrem forte influencia de substituintes
meio alcalino meio alcalino meio alcalino meio alcalino prof.: Ubiracir F. Lima

126
Análises críticas com suporte de pesquisa na literatura.
Um bom ponto de partida é estudar o comportamento de fármacos congêneres Análises críticas com suporte de pesquisa na literatura. TRATADOS K. Florey, Analitycal Profiles of Drug Substances. Academic Press, London. MONOGRAFIAS K.A. Connors, G.L Amidon, V.J. Stella, Chemical Stability of Pharmaceuticals, Wiley, New York, 1986. prof.: Ubiracir F. Lima

127
ε Etapa 2: Informações acerca das propriedades fisico-químicas. λmáx
pKa pka do fármaco x pH da FM KpS Solubilidade na FM Impurezas desconhecidas λmáx ε Detecção Estressar a amostra DAD prof.: Ubiracir F. Lima

128
Etapa 3: Estudos de estresse (Decomposição forçada)
calor 10ºC acima da acelerada umidade 75% ou maior (! ?) pH ácido pH alcalino ICH – Q1A (R2) hidrólise oxidação Não apresenta roteiro prático ! fotólise prof.: Ubiracir F. Lima
 pH ácido. pH alcalino. ICH – Q1A (R2) hidrólise. oxidação. Não apresenta. roteiro prático ! fotólise. prof.: Ubiracir F. Lima.")
129
Roteiros Retirada de alíquotas CCF ou Concentração ou tempo
Degradação hidrolítica HCl / NaOH – 0,1N; 8 h Oxidação H2O2 3-30% Lâmpada fria branca + Fluorescente UV Mínimo de 1,2 milhões de lux h E 200W h/m2 Fotólise Lâmpadas de Xenônio e Haletos metálicos prof.: Ubiracir F. Lima

130
Amostras Condições normais Branco Condições de estresse 4 amostras
H2O Diluidas 1 mg Estresse Neutralizadas H2O + MeOH Resfriadas HPLC prof.: Ubiracir F. Lima

131
Etapa 4: Estudos preliminares para separação das
amostras “estressadas”. [Inicial] 50% Fragmentos iônicos: TRR ~ 15 min. FM DAD pH MeOH/H2O ACN/H2O ODS Amostra multi-componentes MS Otimização gradiente Tampão fluxo coluna temperatura Preparativa LC-MS prof.: Ubiracir F. Lima

132
Degradação do Metronidazol em condições ácidas e sob luz:
Análise inicial; (b) 3 dias; (c) 12 dias; Balanço de massa: área do fármaco x área do PD ? ! prof.: Ubiracir F. Lima
 3 dias; (c) 12 dias; Balanço de massa: área do fármaco x área do PD ! prof.: Ubiracir F. Lima.")
133
Etapa 5: Identificação e caracterização de PD, e
Preparação de padrões. CL-EM Amostra “Estressada” CL-EM-EM CL-RMN Cromatografia Preparativa EM RMN IV prof.: Ubiracir F. Lima

134
Etapa 6: Validação ICH Q2A e Q2B FDA RE 899/03 1ª etapa (fármaco)
2ª etapa (medicamento) especificidade Estender à formulação SIAM seletividade Outras matrizes Limite de detecção e quantificação prof.: Ubiracir F. Lima
 especificidade. Estender à formulação. SIAM. seletividade. Outras matrizes. Limite de detecção. e quantificação. prof.: Ubiracir F. Lima.")
135
Fotoestabilidade – o ICH aponta o teste de luminosidade como uma parte integrante do teste de estresse. Os estudos realizados no fármaco isolado, em solução ou outra formulação, podem gerar informações de características de fotoestabilidade úteis na determinação ou não de estudos adicionais. Se um produto é identificado como fotolábil sob exposição direta, porém é adequadamente protegido por uma embalagem, um estudo “em uso” pode ser necessário para suportar sua validade. Droga parenteral sob infusão por um longo período de tempo Cremes dérmicos prof.: Ubiracir F. Lima

136
Abordagem sistemática para o teste de fotoestabilidade
cobre os seguintes aspectos: Teste na substância ativa Teste no produto fora da embalagem intermediária Teste no produto na embalagem intermediária Teste na embalagem comercial prof.: Ubiracir F. Lima

137
9 - Temperatura Cinética Média (Zonas Climáticas)
USP – temperatura calculada em que o processo de degradação será equivalente àquele resultante da flutuação da temperatura durante o período de estocagem. J. D. Haynes, “Worldwide Virtual Temperatures for Product Stability Testing” , J. Pharm. Sci. 1971 ΔH = calor de ativação; 83,114 KJ/mol R = constante universal dos gases; 8,3144x 10-3 KJ/mol K Tn = média da temperatura de estocagem durante o nésimo período de tempo n = no total de períodos de observação (min. 12) prof.: Ubiracir F. Lima
 prof.: Ubiracir F. Lima.")
138
Aplicações diretas – Zonas climáticas e efeitos cinéticos durante a distribuição
Zonas TCM U.R. % Zona I (temperada) Zona II (subtropical) Zona III (quente e seca) Zona IV (quente e úmida) * WHO Thecnical Report Series, No 863 – 1996 Q1A (R2) fev/03 – zonas climáticas III/IV: 30ºC/65% ICH/FDA – a 252°C / 605%u.r. (zc I/II) prof.: Ubiracir F. Lima
 Zona II (subtropical) Zona III (quente e seca) Zona IV (quente e úmida) 30 75* WHO Thecnical Report Series, No 863 – Q1A (R2) fev/03 – zonas climáticas III/IV: 30ºC/65% ICH/FDA – a 252°C / 605%u.r. (zc I/II) prof.: Ubiracir F. Lima.")
139
prof.: Ubiracir F. Lima

140
Prof.: Ubiracir F. Lima

141
Distribuição - É destacado um novo método leva em consideração o histórico total das condições de estocagem, nas quais determinado produto foi submetido durante o transporte, a estocagem e a distribuição – TCMH. O histórico de exposição é registrado via recurso eletrônico instalado na embalagem do produto, antes da remessa. Quando o produto alcança seu destino, os registros de temperatura e/ou umidade são recolhidos e os valores para TCM são calculados. prof.: Ubiracir F. Lima

142
Utilizando recursos similares, a USP realizou estudos intensivos sobre as condições nas quais os padrões de referência USP e produtos farmacêuticos são expostos durante as fases de expedição. A importância da determinação da TCMH está sendo colocada para produtos sensíveis como vacinas, insulinas e vários produtos biotecnológicos. prof.: Ubiracir F. Lima

143
10 – Equações cinéticas O cálculo do período de tempo, de acordo com a ordem cinética de decomposição, por meio da equação de Arrhenius ou de gráficos tem limitações. substâncias puras e dissolvidas em água. embora cientificamente válido, não se presta para avaliação da estabilidade para formulações contendo outras substâncias dissolvidas e para formulações em que o princípio ativo esteja sob a forma não dissolvida ! Moretto, L.D., Pharma. Technol., 46-8, 1999 prof.: Ubiracir F. Lima

144
Estabilidade Equações cinéticas
Velocidades da reações químicas: V α [F] [R] V = [F] [R] Determina-se uma constante de proporcionalidade: K = K [F] [R] * Equação de 2a ordem prof.: Ubiracir F. Lima

145
Estabilidade Equações cinéticas
Ordem de reação: soma dos expoentes das espécies envolvidas nas reações. Equação de 1a ordem = K [F] Equação de ordem zero = K prof.: Ubiracir F. Lima

146
Estabilidade Equações cinéticas
* limitações Orbitais moleculares: densidade eletrônica, estequiometria das reações Mecanismo das reações: número de colisões prof.: Ubiracir F. Lima

147
Estabilidade Equações cinéticas
prof.: Ubiracir F. Lima

148
Estabilidade Equações cinéticas
7.1-Determinação dos prazos de validade a partir das equações cinéticas Reações de 1a ordem = K [F] = - K dt = -K dt ln [F] = ln [F]o – K t prof.: Ubiracir F. Lima

149
Estabilidade Equações cinéticas
O prazo de validade do fármaco é observado quando [F] = 0,9[F]o ln 0,9[F]o = ln[F]o – Kt90 prof.: Ubiracir F. Lima

150
Estabilidade Equações cinéticas
Em pH 2,5, a constante de velocidade (K) para degradação do ácido acetil salicílico na solução de aspirina é de 5 x 10 –7 s-1 a 25°C. t90 = 0,105 / 5 x = 2,1 x 105s = 2 dias prof.: Ubiracir F. Lima
 para degradação do ácido acetil salicílico na solução de aspirina é de 5 x 10 –7 s-1 a 25°C. t90 = 0,105 / 5 x = 2,1 x 105s = 2 dias. prof.: Ubiracir F. Lima.")
151
Estabilidade Equações cinéticas
Reações de ordem zero – = K d[F] = - K dt d[F] = -K dt [F] – [F]o = - k t quando [F] = 0,9[F]o ,9[F]o – [F] = - Kt prof.: Ubiracir F. Lima

152
Estabilidade Equações cinéticas
A solubilidade da aspirina em pH 2,5 é de 0,33g/100mL. Formulando este fármaco como suspensão de 13g/100mL teremos K = 1,65 x 10-7 g/ mL . s A concentração da aspirina é constante. A velocidade da reação independe da concentração inicial Equação de ordem zero. t90 = = 7,9 x 106 s = dias prof.: Ubiracir F. Lima

153
Estabilidade Equações cinéticas
7.2- Efeitos da temperatura – Eq. De Arrhenius. O aumento da temperatura, aumenta o número de colisões e conseqüentemente a velocidade das reações. É determina uma relação exponencial da constante da velocidade com a temperatura. Eq. de Arrhenius K = A e –Ea/Rt log K = log A – Ea/2,303 RT A = constante de freqüência das colisões Ea = energia de ativação R = constante universal dos gases T = temperatura absoluta prof.: Ubiracir F. Lima

154
Estabilidade Equações cinéticas
Para determinar o prazo de validade (t90), deve-se obter a constante de velocidade (K) para a temperatura em que deseja-se armazenar o produto. a) Armazenando os produtos em condições de estabilidade acelerada, obtém-se valores de K em temperaturas diferentes. se o gráfico [ F ] x T resultar em uma reta, trata-se de reação de ordem zero. se o gráfico log [ F ] x T resultar em uma reta, trata-se de reação de 1a ordem. prof.: Ubiracir F. Lima
, deve-se obter a constante de velocidade (K) para a temperatura em que deseja-se armazenar o produto. a) Armazenando os produtos em condições de estabilidade acelerada, obtém-se valores de K em temperaturas diferentes. se o gráfico [ F ] x T resultar em uma reta, trata-se de reação de ordem zero. se o gráfico log [ F ] x T resultar em uma reta, trata-se de reação de 1a ordem. prof.: Ubiracir F. Lima.")
155
Estabilidade Equações cinéticas
b) por meio da eq. de Arrhenius, extrapola-se para determinação de K na temperatura desejada. Lembrando: ordem zero t90 = 0,1 [ F ]o / K 1a ordem t90 = 0,105 / K prof.: Ubiracir F. Lima
 por meio da eq. de Arrhenius, extrapola-se para determinação de K na temperatura desejada. Lembrando: ordem zero t90 = 0,1 [ F ]o / K. 1a ordem t90 = 0,105 / K. prof.: Ubiracir F. Lima.")
156
RESOLUÇÃO - RE Nº. 1, DE 29 DE JULHO DE 2005.
O Diretor-Presidente da Agência Nacional de Vigilância Sanitária, no uso de suas atribuições e tendo em vista o disposto no art. 111, inciso II, alínea "a" do Regimento Interno da ANVISA aprovado pela Portaria nº 593, de 25 de agosto de 2000, republicada em 22 de dezembro de 2000, resolve: Art. 1º Autorizar ad referendum, a publicação do Guia para a Realização de Estudos de Estabilidade, em anexo. Art. 2° Fica revogada a Resolução - RE n° 398, de 12 de novembro de 2004, publicada no Diário Oficial da União de 16 de novembro de 2004. Art. 3° Esta Resolução entra em vigor na data de sua publicação.

157
GUIA PARA A REALIZAÇÃO DE ESTUDOS DE ESTABILIDADE
A estabilidade de produtos farmacêuticos depende de fatores ambientais como temperatura, umidade e luz, e de outros relacionados ao próprio produto como propriedades físicas e químicas de substâncias ativas e excipientes farmacêuticos, forma farmacêutica e sua composição, processo de fabricação, tipo e propriedades dos materiais de embalagem. APLICABILIDADE Guia para realização dos testes de estabilidade de produtos farmacêuticos a fim de prever, determinar ou acompanhar o seu prazo de validade.

158
1. DEFINIÇÕES ESTUDO DE ESTABILIDADE ACELERADO Estudo projetado para acelerar a degradação química e/ou mudanças físicas de um produto farmacêutico em condições forçadas de armazenamento. Os dados assim obtidos, juntamente com aqueles derivados dos estudos de longa duração, podem ser usados para avaliar efeitos químicos e físicos prolongados em condições não aceleradas e para avaliar o impacto de curtas exposições a condições fora daquelas estabelecidas no rótulo do produto, que podem ocorrer durante o transporte. ESTUDO DE ESTABILIDADE DE ACOMPANHAMENTO Estudo realizado para verificar que o produto farmacêutico mantém suas características físicas, químicas, biológicas, e microbiológicas conforme os resultados obtidos nos estudos de estabilidade de longa duração.

159
ESTUDO DE ESTABILIDADE DE LONGA DURAÇÃO
Estudo projetado para verificação das características físicas, químicas, biológicas e microbiológicas de um produto farmacêutico durante e, opcionalmente, depois do prazo de validade esperado. Os resultados são usados para estabelecer ou confirmar o prazo de validade e recomendar as condições de armazenamento. LOTE Quantidade de um produto obtido em um único processo ou série de processos, cujas características essenciais são a homogeneidade e qualidade dentro dos limites especificados. LOTE EM ESCALA PILOTO Um lote de produto farmacêutico produzido por um processo totalmente representativo simulando o lote de produção industrial e estabelecido por uma quantidade mínima equivalente a 10% do lote industrial previsto, ou quantidade equivalente à capacidade mínima do equipamento industrial a ser utilizado.

160
PRAZO DE VALIDADE Data limite para a utilização de um produto farmacêutico definida pelo fabricante, com base nos seus respectivos testes de estabilidade, mantidas as condições de armazenamento e transporte estabelecidos. TESTE DE ESTABILIDADE Conjunto de testes projetados para obter informações sobre a estabilidade de produtos farmacêuticos visando definir seu prazo de validade e período de utilização em embalagem e condições de armazenamento especificadas. 2. DISPOSIÇÕES GERAIS 2.1 O prazo de validade de um produto a ser comercializado no Brasil é determinado por um estudo de estabilidade de longa duração de acordo com os parâmetros definidos em tabela abaixo. Por ocasião do registro poderá ser concedido um prazo de validade provisório de 24 meses se aprovado o relatório de estudo de estabilidade de longa duração de 12 meses ou relatório de estudo de estabilidade acelerado de 6 meses acompanhado dos resultados preliminares do estudo de longa duração com, conforme parâmetros definidos em tabela abaixo.

161
Forma Farm. Cond. de armaz. Embalagem Acelerado Longa Duração
Sólido °C -30°C Semi-permeável ºC +/-2 / 75% +/-5UR ºC+/- 2 / 75% +/-5 UR Sólido °C -30°C Impermeável ºC +/ ºC+/- 2 Semi-sólido °C -30°C Semi-permeável ºC +/-2 / 75% +/-5UR ºC+/- 2 / 75% +/-5 UR Semi-sólido °C -30°C Impermeável ºC +/ ºC+/- 2 Líquidos °C -30°C Semi-permeável ºC +/-2 / 75% +/-5UR ºC+/- 2 / 75% +/-5 UR Líquidos °C -30°C Impermeável ºC +/ ºC+/- 2 Gases °C -30°C Impermeável ºC +/ ºC+/- 2 Todas as formas farmacêuticas 2°C - 8°C Impermeável ºC +/- 2º ºC +/- 3º 2°C - 8°C Semi-permeável ºC +/- 2ºC / 60 % +/-5% ºC +/- 3º -20 °C Todas ºC +/- 5º ºC +/- 5º

162
* Qualquer recomendação de armazenamento em temperatura dentro destas faixas deve constar de bulas e rótulos. A temperatura recomendada não exime de que os testes de estabilidade sejam realizados com as temperaturas definidas nas duas últimas colunas da tabela. ** Os valores de temperatura e umidade são fixos e as variações são inerentes às oscilações esperadas pela câmara climática e por eventuais aberturas para retirada ou colocação de material. *** Líquidos e semi-sólidos de base aquosa devem realizar o estudo com umidade a 25% UR ou 75% UR. Caso se opte por 75% UR, o valor da perda de peso deverá ser multiplicado por 3,0. 2.2. O prazo de validade deve ser confirmado mediante a apresentação de um estudo de estabilidade de longa duração de 24 meses de duração, protocolado na forma de complementação de informações ao processo. A presença desta documentação no processo é necessária para a renovação do registro.

163
2.3. O estudo de estabilidade deve ser executado com o produto farmacêutico em sua embalagem primária. 2.4. Os produtos importados a granel devem descrever nos seus rótulos a data de fabricação, a validade e a condição de armazenamento até a execução da embalagem primária para serem liberados pela autoridade sanitária de portos e aeroportos. O estudo será avaliado durante a inspeção na empresa fabricante. 2.5. Para produtos importados, os estudos de estabilidade podem ser realizados no exterior de acordo com os parâmetros definidos nesta Resolução. Nos caso de produtos importados a granel, o prazo de validade deve levar em consideração o tempo máximo de armazenamento até a execução da embalagem primária. 2.6. Para produtos importados, a granel ou em embalagem primária, os estudos de estabilidade de acompanhamento devem ser realizados em solo brasileiro de acordo com os parâmetros definidos nesta Resolução.

164
2.7. Estudos adicionais, tais como fotoestabilidade que se façam pertinentes de acordo com as propriedades do produto em questão, poderão ser necessárias para a comprovação da estabilidade de produtos farmacêuticos. Nestes casos sugerimos seguir recomendação técnica disponível no portal da Anvisa. A não apresentação de estudo de fotoestabilidade deve vir acompanhada de justificativa técnica com evidência científica de que o(s) ativos(s) não sofre(m) degradação em presença de luz ou de que a embalagem primária não permite a passagem de luz. 2.8. É facultado utilizar modelos reduzidos de plano de estudo de estabilidade, baseados nos princípios estabelecidos na recomendação técnica disponível no portal da Anvisa. 2.9. Todo relatório de estudo de estabilidade, independente da forma farmacêutica, deve apresentar as seguintes informações ou justificativa técnica de ausência:

165
Descrição do produto com respectiva especificação da embalagem primária
Número do lote para cada lote envolvido no estudo Descrição do fabricante dos princípios ativos utilizados Aparência Plano de estudo: material, métodos (desenho) e cronograma. Data de início do estudo Teor do princípio ativo e método analítico correspondente Quantificação de produtos de degradação e método analítico correspondente Limites microbianos Para toda a forma farmacêutica sólida a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: Dissolução Dureza Para as formas farmacêuticas líquídas e semi-sólidas, a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: pH Sedimentação pós agitação em suspensões Claridade em soluções Separação de fase em emulsões e cremes Perda de peso em produtos de base aquosa
 e cronograma. Data de início do estudo Teor do princípio ativo e método analítico correspondente Quantificação de produtos de degradação e método analítico correspondente Limites microbianos Para toda a forma farmacêutica sólida a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: Dissolução Dureza Para as formas farmacêuticas líquídas e semi-sólidas, a empresa deve acrescentar as seguintes informações ou justificativa técnica de ausência: pH Sedimentação pós agitação em suspensões Claridade em soluções Separação de fase em emulsões e cremes Perda de peso em produtos de base aquosa")
166
2.10. Para fins de prazo de validade provisório de 24 meses será aprovado o relatório de estabilidade acelerado ou de longa duração de 12 meses que apresentar variação menor ou igual a 5,0% do valor de análise da liberação do lote, mantidas as demais especificações. Caso as variações de doseamento estejam entre 5,1% e 10,0% no estudo de estabilidade acelerado, o prazo de validade provisório será reduzido à metade, ou seja, será de 12 meses. O doseamento no momento zero não pode ultrapassar as especificações do produto de acordo com farmacopéias reconhecidas pela Anvisa ou, na ausência de informação farmacopeica, com método validado de acordo com o Guia para validação de métodos analíticos e bioanalíticos. Caso a especificação farmacopeica e/ou proveniente de método validado permitir que o momento zero seja acima de 10% do declarado a variação da queda será analisada caso a caso. 2.11. Para fins de prazo de validade definitivo, somente será aprovado o relatório de estabilidade que apresentar a variação do doseamento dos princípios ativos dentro das especificações farmacopeicas e/ou proveniente de método validado do produto de acordo com o Guia para validação de métodos analíticos e bioanalíticos, e mantidas as demais características do produto.

167
2.12. Em caso de produtos que requeiram reconstituição ou diluição deve-se apresentar informações iniciais e finais que comprovem o período de utilização pelo qual o produto mantém a sua estabilidade depois da reconstituição, nas condições de armazenamento determinadas. Os estudos devem ser conduzidos utilizando o diluente especificado para reconstituição do produto farmacêutico. Se existir a opção de mais de um diluente, o estudo deve ser conduzido com aquele que apresente o produto farmacêutico reconstituído menos estável. 2.13. Em caso de comprimidos efervescentes deve-se apresentar informações iniciais e finais que comprovem o período de utilização pelo qual o produto remanescente mantém a sua estabilidade depois da abertura da embalagem primária nas condições de armazenamento determinadas. Estes estudos devem ser realizados com os parâmetros e testes definidos por esta Resolução. 2.14. Excepcionalmente, para os produtos cujos cuidados de conservação sejam inferiores a 25°C e de uso exclusivo em hospitais e clínicas médicas, serão aceitos estudos de estabilidade nas condições especificadas para Zona II (25°C/60%UR), desde que seja devidamente comprovado que o produto não suporta as condições estabelecidas nesta Resolução. Entretanto o titular do registro do produto deve assegurar a conservação recomendada durante o transporte e a distribuição.
, desde que seja devidamente comprovado que o produto não suporta as condições estabelecidas nesta Resolução. Entretanto o titular do registro do produto deve assegurar a conservação recomendada durante o transporte e a distribuição.")
168
3. SELEÇÃO DE LOTES 3.1. Para fins de registro e alterações pós-registro, nos estudos de estabilidade acelerado e longa duração: um ou três lotes, de acordo com as normas legais e regulamentares pertinentes. 3.2. Os lotes a serem amostrados devem ser representativos do processo de fabricação, tanto em escala piloto quanto escala industrial. 3.3. Para os produtos cuja concentração do princípio ativo esteja na ordem de dosagem abaixo de 0.99 miligramas por unidade posológica, não serão permitidos lotes pilotos com quantitativos diferentes dos lotes industriais. Não aplicável a soluções. 3.4. Os estudos de acompanhamento deverão ser realizados nas condições climáticas preconizadas neste Guia. A amostragem deve seguir os parâmetros abaixo descritos:

169
a) Um lote anual, para produção acima de 15 lotes/ano.
b) Um lote a cada 2 anos, produção abaixo ou igual de 15 lotes/ano. c) Para produtos com diferentes concentrações e formulações proporcionais, poderá ser utilizado como critério de escolha, aquele que apresentar o maior número de lotes produzidos ao ano. 3.5. O estudo de acompanhamento somente poderá ser realizado se o produto não sofrer nenhuma alteração após a conclusão do estudo de estabilidade de longa duração. Caso ocorra qualquer alteração no produto deverá ser realizado novo estudo de estabilidade de longa duração conforme preconizado neste Guia. 4. FREQÜÊNCIA DOS TESTES 4.1. Estudo acelerado: 0, 3 e 6 meses para doseamento, quantificação de produtos de degradação, dissolução (quando aplicável) e pH (quando aplicável). Para as demais provas apresentar estudo aos 6 meses comparativo ao momento zero.
 Um lote a cada 2 anos, produção abaixo ou igual de 15 lotes/ano. c) Para produtos com diferentes concentrações e formulações proporcionais, poderá ser utilizado como critério de escolha, aquele que apresentar o maior número de lotes produzidos ao ano O estudo de acompanhamento somente poderá ser realizado se o produto não sofrer nenhuma alteração após a conclusão do estudo de estabilidade de longa duração. Caso ocorra qualquer alteração no produto deverá ser realizado novo estudo de estabilidade de longa duração conforme preconizado neste Guia. 4. FREQÜÊNCIA DOS TESTES 4.1. Estudo acelerado: 0, 3 e 6 meses para doseamento, quantificação de produtos de degradação, dissolução (quando aplicável) e pH (quando aplicável). Para as demais provas apresentar estudo aos 6 meses comparativo ao momento zero.")
170
4.2. Estudo de longa duração: 0, 3, 6, 9, 12, 18, 24 meses para doseamento, quantificação de produtos de degradação, dissolução (quando aplicável) e pH (quando aplicável). Para as demais provas, apresentar estudo no prazo de validade requerido comparativo ao momento zero. 4.3. Estudo de acompanhamento: a cada 12 meses deverão ser realizados todos os testes de um relatório de estudo de estabilidade, relatório que deve ser disponibilizado no momento da inspeção. 5. DA ADEQUAÇÃO 5.1. É obrigatória a apresentação de estudos de estabilidade no momento da primeira renovação de registro após a publicação desta Resolução caso este estudo não conste do dossiê do registro, mesmo que conduzidos de acordo com os parâmetros vigentes quando do início dos estudos. 5.2. A Anvisa aceitará até 31 de julho de 2007, no momento do registro, pós-registro ou da renovação de registro, estudos de estabilidade de longa duração que já estejam em andamento com o parâmetro de umidade abaixo de 75%. Entretanto as câmaras climáticas devem ser re-qualificadas para umidade de 75% a partir da data de publicação desta Resolução. Fica a critério da empresa reiniciar ou não estes estudos.

171
5.3. Caso os estudos de estabilidade de longa duração tenham sido realizados somente com parâmetros de umidade distintos do definido nesta Resolução, as empresas deverão na primeira renovação de registro após 1 de agosto de 2007, apresentar estudos de estabilidade de acompanhamento em um lote, de acordo com esta Resolução. Para produtos que atendam a esta circunstância, o momento da renovação é entre 1 de agosto de 2007 e 31 de julho de 2008 e já tenham validade igual ou superior a 36 meses, é possível aprovar um período de validade de 36 meses com a apresentação de estudos de no mínimo de 24 meses. 5.4. Caso os estudos de estabilidade de longa duração tenham sido realizados com parâmetros de temperatura e umidade distintos do definido nesta Resolução, as empresas deverão na primeira renovação de registro após 1 de agosto de 2007, apresentar estudos de estabilidade de longa duração de 12 meses, ou estudo acelerado de 6 meses acompanhado do respectivo estudo de longa duração de acordo com esta Resolução. Caso as condições de estabilidade não forem comprovadas na submissão da renovação de registro, a empresa deverá solicitar suspensão de comercialização para manter o registro, caso contrário o registro não será renovado.

172
5.5. Casos os estudos de longa duração, realizados através das condições desta Resolução, comprovem um prazo de validade menor que o estabelecido no registro do produto, a empresa deverá imediata e provisoriamente implementar e solicitar alteração pós-registro para alteração de prazo de validade com base nos dados obtidos.

173
Obrigado !

Apresentações semelhantes
 – Lápides 1, 2, 3» «nomes gravados, 21 de Agosto de 2008» «Ultramar.TerraWeb»>")


















