Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Disciplina 5397 – Eletroquímica e Corrosão
Universidade Federal de Goiás Instituto de Química Disciplina 5397 – Eletroquímica e Corrosão

2
Transferência de elétron Butler-Vomer Polarização eletrótica Transporte de massa

3
Fundamentos da cinética e dos mecanismos das reações de eletrodos
Caso mais simples: Transferência de um elétron sem transformação química ( formação ou quebra de uma ligação química) Exemplo: Fe3+(aq) + e- Fe2+(aq) Para essa reação ocorre: 1 – Difusão da espécie até a superfície do eletrodo 2 – Rearranjo da atmosfera iônica 3 – Reorientação dos dipolos do solvente 4 – Alteração entre as distancias dos íons central e os ligados 5 – Transferência do elétron 6 – Relaxação no sentido inverso
 Exemplo: Fe3+(aq) + e- Fe2+(aq) Para essa reação ocorre: 1 – Difusão da espécie até a superfície do eletrodo. 2 – Rearranjo da atmosfera iônica. 3 – Reorientação dos dipolos do solvente. 4 – Alteração entre as distancias dos íons central e os ligados. 5 – Transferência do elétron. 6 – Relaxação no sentido inverso.")
4
Modelos da dupla camada elétrica
Generalizando: O + ne- R Ka O + ne- R Kc Kc e Ka incluem os passos de difusão, reorganização, adsorção e a transferência do elétron Mecanismo similar ocorre em solução homogênea, entretanto, sítio de transferência não é o eletrodo é a outra espécie química.

5
A velocidade da reação é dada por :
A velocidade da reação é dada por : Velocidade catódica Velocidade anódica Vc = KcCo VR = KRCo e Co e Cr = concentração das espécies O e R 5

6
Pela equação de Arrhenius: K = A exp (-Ea / RT)
Onde Ea é a Energia de ativação, e tem unidades de energia e A é um fator de frequência O valor de Ea pode ser obtido pelo coeficiente angular da reta: ln k vs 1/T Podemos usar Ea como sendo a Energia Interna padrão, que vai do valor mínimo para o máximo – o estado de transição ou complexo ativado Então Ea é a Energia Interna padrão de ativação Com isso, a entalpia padrão de ativação é: H = U + pV ΔH = Δ Ea + Δ(pV) Como a reação acontece em fase líquida Δ(pV) tem valor desprezível Então: ΔH* Δ Ea E a equação de Arrhenius pode ser escrita como : K = A exp (- ΔH* / RT) 6
 Como a reação acontece em fase líquida Δ(pV) tem valor desprezível. Então: ΔH* Δ Ea. E a equação de Arrhenius pode ser escrita como : K = A exp (- ΔH* / RT) 6.")
7
K = A exp (- ΔH* / RT) Podemos considerar também o fator A como A = A’ exp (ΔS* / R) isso porque, o exponencial envolvendo a entropia padrão de ativação, ΔS*, é uma constante adimensional. Substituindo na equação de Arrehenius K = A exp (- ΔH* / RT) A = A’ exp (ΔS* / R)
 A = A’ exp (ΔS* / R)")
8
mmc = RT Sabemos que ΔG = ΔH – TΔS A equação de Arrhenius fica: Onde ΔG* é a energia livre padrão de ativação Como o potencial do eletrodo pode interferir em ΔG* Considerando a reação O + ne- R Velocidade catótica Velocidade anódica Vc = KcCo VR = KRCo

9
Como o potencial do eletrodo pode interferir em ΔG*
Considerando a reação O + ne- R Velocidade catódica Velocidade anódica Vc = KcCo VR = KRCR Substituindo esses valores na equação de Arrhenius onde k1 e k2 são fatores de frequência de colisão dos reagentes e produtos com a superfície do eletrodo v = kC vR = kACR KA = v/CR vc = kcCo Kc = v/Co

10
i = ic – iA Aplicando a lei de Faraday: I = F v v = i/F
Energia de ativação da reação R O Energia de ativação da reação O R Aproximadamente = distancia do eletrodo Aplicando a lei de Faraday: I = F v v = i/F O que mede no amperímetro i Corrente que flui através do sistema: i = ic – iA Adotando que a corrente catódica tem um valor positivo

11
O que se mede efetivamente é i
i = ic – iA Se i = 0; ic = iA Então, no eletrodo há um equilíbrio Ou seja: i = ic – iA

12
i = ic – iA Substituindo na equação: A energia de ativação é um dos pontos mais importantes que diferenciam a cinética química da cinética eletroquímica No caso da reação eletroquímica: O + ne- R Para a reação acontecer, a espécie O deve se aproximar do eletrodo No instante antes da transferência e ainda está vinculado ao metal (eletrodo)
")
13
Probabilidade praticamente nula
No instante em que a espécie O está na superfície do eletrodo, e o elétron é transferido para O. O se transforma em R e- R Pela mecânica quântica: I – Existe a probabilidade de e- estar no nível e Fermi (banda de condução mais energético) no metal. Se O absorve o elétron transforma-se em R e isso determina a velocidade da reação catódica Vc = KcCo II - Transferência do elétron só é possível se: i) O apresentar um orbital desocupado, ii) Orbital com energia igual a do nível de Fermi do metal (para que não haja radiação de energia) Probabilidade praticamente nula Reação parece impossível mas acontece
 no metal. Se O absorve o elétron transforma-se em R e isso determina a velocidade da reação catódica. Vc = KcCo. II - Transferência do elétron só é possível se: i) O apresentar um orbital desocupado, ii) Orbital com energia igual a do nível de Fermi do metal. (para que não haja radiação de energia) Probabilidade praticamente nula. Reação parece impossível mas acontece.")
14
Explicação Espécie O não está estática na superfície do eletrodo: Em solução há: Agitação térmica Choque com solvente Colisão com o eletrodo Efeito do potencial da dupla camada elétrica Esses efeitos distorcem a molécula O, alteram a camada de hidratação da espécie O, ou seja, torna a espécie ativa, isto é: Orbitais moleculares com elevada energia: O O* Com isso, o elétron é transferido por efeito túnel Ou seja, a energia de ativação trata-se da distorção da molécula da espécie que vai receber o elétron, a transição do elétron não tem nenhuma barreira energética. A transferência do elétron não pode ocorrer em diferentes níveis de energia, porque não é um transferência com irradiação de energia.

15
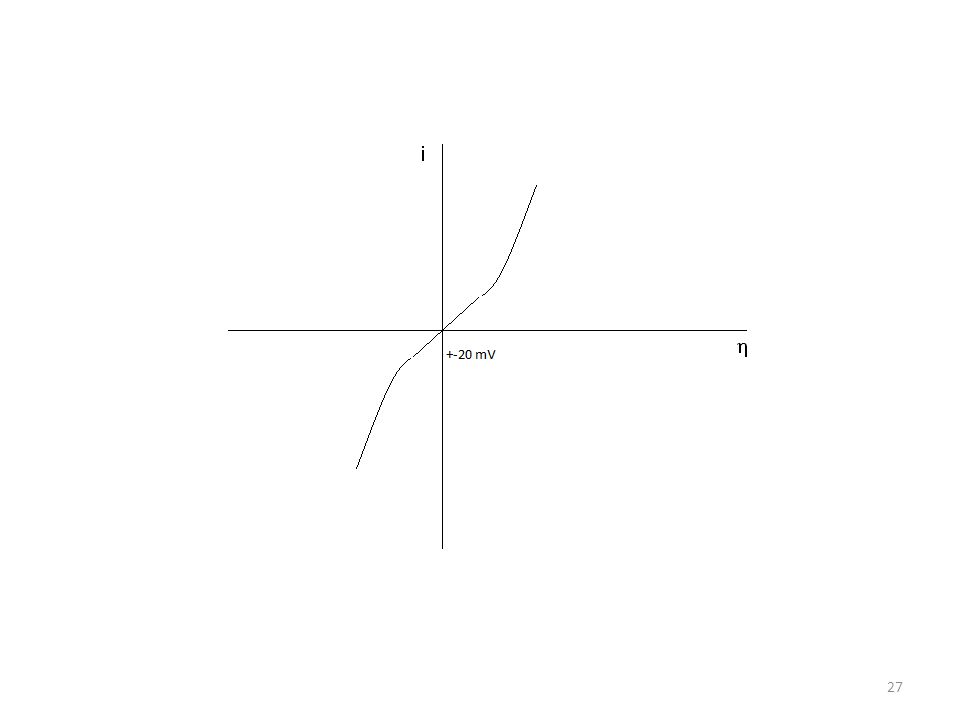
Na interface eletrodo/solução há intenso campo elétrico Δ φ e diferença de potencial eletrodo/solução é alterada externamente com o potenciostato Δ φ = Δ φ e + η

16
Δ φe = diferença de potencial existente (no equilíbrio)
Lembrando que a interface eletrodo/solução possui intenso campo elétrico Δ φ Essa diferença de potencial Δφ aparece naturalmente na superfície do eletrodo, mesmo quando a corrente que flui na interface é zero e diferença de potencial eletrodo/solução é alterada externamente com o potenciostato Δ φ = Δ φ e + η Δ φe = diferença de potencial existente (no equilíbrio) η = contribuição externa sobrepotencial (sobretensão) A contribuição externa pode tornar o eletrodo positivo ou negativo, assim pode-se escrever: η = E - Ee Utilizando uma célula eletroquímica contendo três eletrodos
 η = contribuição externa sobrepotencial (sobretensão) A contribuição externa pode tornar o eletrodo positivo ou negativo, assim pode-se escrever: η = E - Ee. Utilizando uma célula eletroquímica contendo três eletrodos.")
17
Célula eletroquímica com três eletrodos
Eletrodo de trabalho Eletrodo de referência Contra eletrodo Fluxo de corrente (i) Polarização (E)
 Polarização (E)")
18
i = ic – iA Substituindo na equação: Considerando a reação: O + ne- R Toda reação de redução tem um potencial Eredox = fixo . Entretanto o potencial do eletrodo pode ser alterado, o que modifica o nível eletrônico ocupado mais elevado no eletrodo (nível de Fermi, EF) O potencial aplicado ao eletrodo pode mudar a direção da transferência do elétron Cátodo ocorre redução espécie ganha elétrons do eletrodo; e no Ânodo ocorre oxidação espécies perdem elétrons para o eletrodo
 O potencial aplicado ao eletrodo pode mudar a direção da transferência do elétron. Cátodo ocorre redução espécie ganha elétrons do eletrodo; e no. Ânodo ocorre oxidação espécies perdem elétrons para o eletrodo.")
19
A energia de ativação de uma reação eletroquímica tem duas contribuições:
Para um mol de elétrons, zie = F e Δ φ1 é proporcional à diferença de potencial da interface, Sendo uma constante de proporcionalidade igual a , temos: Δ φ = Δ φ e + η Substituindo esses valores na equação de i

20
Δ φ = Δ φ e + η É o fator de simetria, um número adimensional que varia de 0 a 1 O valor de é dado por 1- x/X – Na maioria dos processos eletroquímicos = 0,5

21
i 0 = densidade de corrente de troca do sistema
Essa equação pode ser simplifica: Quando η = 0 o sistema encontra-se em equilíbrio, Como consequência i = 0 então i = ic = iA Podemos definir i o como i 0 = densidade de corrente de troca do sistema

22
Onde io é uma grandeza característica da reação e do material eletródico usado Com base na equação de io, a equação de i fica: Essa equação pode ser rearranjada para: Equação de Butler-Volmer Sistemas no equilíbrio i = 0 Equação de Nernst Sistemas fora equilíbrio i 0 Equação de Butler-Volmer

23
Δ φ e = Δ φo e – RT/F ln (Co / CR)
Análise da equação de Butler-Volmer Sistemas em equilíbrio η =0 Sistemas em equilíbrio i =0 i 0= F Co K1 exp(- ΔGo≠o +F α (Δ φ -η) / RT) – = F CR K2 exp(- ΔGo≠o -F (1-α) (Δ φ -η) / RT) i 0= F Co K1 exp(- ΔGo≠o +F α (Δ φ -η) / RT) – = F CR K2 exp(- ΔGo≠o -F (1-α) (Δ φ -η) / RT) F Co K1 exp(- ΔGo≠o +F α (Δ φ -η) / RT) = F CR K2 exp(- ΔGo≠o -F (1-α) (Δ φ -η) / RT) Que pode ser reescrita como: Δ φ e = Δ φo e – RT/F ln (Co / CR) Equação de Nernst
 / RT) – = F CR K2 exp(- ΔGo≠o -F (1-α) (Δ φ -η) / RT) i 0= F Co K1 exp(- ΔGo≠o +F α (Δ φ -η) / RT) – = F CR K2 exp(- ΔGo≠o -F (1-α) (Δ φ -η) / RT) F Co K1 exp(- ΔGo≠o +F α (Δ φ -η) / RT) = F CR K2 exp(- ΔGo≠o -F (1-α) (Δ φ -η) / RT) Que pode ser reescrita como: Δ φ e = Δ φo e – RT/F ln (Co / CR) Equação de Nernst.")
24
i = i 0 [ exp(- α F η) / RT) – exp ( (1-α) F η) / RT)
Análise da equação de Butler-Volmer Sistemas próximo ao equilíbrio η 0 Expoentes da equação são pequenos – os termos exponenciais pode ser expandidos em série e ± x (x 0) = 1 ± x ± x2/2! ± x3/3! 1 ± x i = i 0 [ exp(- α F η) / RT) – exp ( (1-α) F η) / RT) A equação fica: i = - i 0 F η / RT Densidade de corrente varia linearmente com o potencial
 = 1 ± x ± x2/2! ± x3/3! 1 ± x. i = i 0 [ exp(- α F η) / RT) – exp ( (1-α) F η) / RT) A equação fica: i = - i 0 F η / RT. Densidade de corrente varia linearmente com o potencial.")
25
Análise da equação de Butler-Volmer
Sistemas afastados do equilíbrio η<< 0 catódica exp(- αF η) / RT >> exp (1-α) F η) / RT assim i = io exp(- α F η) / RT rearranjada η = RT/αF ln io – RT/ αF ln i η = b – a lni Equação de Tafel
 / RT >> exp (1-α) F η) / RT. assim. i = io exp(- α F η) / RT. rearranjada. η = RT/αF ln io – RT/ αF ln i. η = b – a lni. Equação de Tafel.")
26
Análise da equação de Butler-Volmer
Sistemas afastados do equilíbrio η>> 0 anódica exp(- α F η) / RT << exp (1-α) F η) / RT η = -RT/ (1-α) F ln |io| + RT/ (1-α) F ln |i| ou
 / RT << exp (1-α) F η) / RT. η = -RT/ (1-α) F ln |io| + RT/ (1-α) F ln |i| ou.")
28
Polarização eletródica
Quando os sistemas eletroquímicos estão fora do equilíbrio ( i ≠ 0) o potencial dos eletrodos (E) é diferente do valo do potencial no equilíbrio E(i ≠ 0) ≠ E A diferença deste potencial é maior quanto maior a corrente que atravessa a interface eletrodo/solução E = f(i) O potencial do eletrodo depende da corrente que circula no sistema. A diferença entre E e Eeq é chamada de polarização eletrótica
 o potencial dos eletrodos (E) é diferente do valo do potencial no equilíbrio. E(i ≠ 0) ≠ E. A diferença deste potencial é maior quanto maior a corrente que atravessa a interface eletrodo/solução. E = f(i) O potencial do eletrodo depende da corrente que circula no sistema. A diferença entre E e Eeq é chamada de polarização eletrótica.")
29
Principais etapas para ocorrer a reação eletroquímica:
1 – a espécie tem que se aproximar da interface eletrodo/solução (onde ocorre a reação) 2 – na interface deve ocorrer a transferência de carga reagente transforma em produto 3 – simultaneamente, a carga envolvida no processo deve ser transportada ao outro eletrodo (garantindo a eletroneutalidade da solução) Se as 3 etapas forem rápidas a polarização eletródica é pequena. É comum que uma ou mais destas etapas sejam lentas ( etapa determinante da velocidade da reação)
 2 – na interface deve ocorrer a transferência de carga reagente transforma em produto. 3 – simultaneamente, a carga envolvida no processo deve ser transportada ao outro eletrodo (garantindo a eletroneutalidade da solução) Se as 3 etapas forem rápidas a polarização eletródica é pequena. É comum que uma ou mais destas etapas sejam lentas ( etapa determinante da velocidade da reação)")
30
1 – Se o reagente estiver em baixa concentração e/ou a corrente elevada tende a um esgotamento de reagente na superfície do eletrodo. Velocidade da reação determinada pela velocidade de chegada de espécies reagentes na superfície do eletrodo. O potencial do eletrodo muda em relação ao potencial no estado de equilíbrio – neste caso – chamado polarização por transporte de massa

31
2 – A concentração é suficientemente alta ou a corrente é baixa
2 – A concentração é suficientemente alta ou a corrente é baixa. A transferência do elétron do eletrodo para o reagente (ou vice-versa) é limitada por uma barreira de energia de ativação. Se este fenômeno é a etapa determinante da velocidade da reação ocorre a polarização por ativação.
 é limitada por uma barreira de energia de ativação. Se este fenômeno é a etapa determinante da velocidade da reação ocorre a polarização por ativação.")
32
3 – Concentração iônica baixa (ou condutividade iônica baixa) dificuldade em manter eletroneutralidade da solução diminui a velocidade do processo global. Este fenômeno é governado pela lei de condutância eletrolítica (lei de Ohm) polarização por queda Ohmica.
 polarização por queda Ohmica..")
33
Polarização por ativação Na interface eletrodo/solução há intenso campo elétrico Δ φ e diferença de potencial eletrodo/solução é alterada externamente com o potenciostato Δ φ = Δ φ e + η

34
Polarização por queda Ohmica
Concentração iônica baixa (ou condutividade iônica baixa) dificuldade em manter eletroneutralidade da solução diminui a velocidade do processo global. Este fenômeno é governado pela lei de condutância eletrolítica (lei de Ohm). A polarização por queda Ôhmica pode ser minimizada mas nunca completamente eliminada existe, por menor que seja, uma resistência à passagem de corrente em qualquer eletrólito. Considerando o sistema eletroquímico de três eletrodos. ΔV = ΔVo – IR, ΔVo i = 0 Mas E = E rev + η E = E rev – RI
 dificuldade em manter eletroneutralidade da solução diminui a velocidade do processo global. Este fenômeno é governado pela lei de condutância eletrolítica (lei de Ohm). A polarização por queda Ôhmica pode ser minimizada mas nunca completamente eliminada existe, por menor que seja, uma resistência à passagem de corrente em qualquer eletrólito. Considerando o sistema eletroquímico de três eletrodos. ΔV = ΔVo – IR, ΔVo i = 0. Mas E = E rev + η. E = E rev – RI.")
35
Com isso sistemas espontâneos (pilhas) efeito ôhmico leva a uma diminuição de voltagem e consequentemente diminuição de potência de saída. sistemas não espontâneos (eletrolisadores) observa-se uma aumento na voltagem e na potência de entrada. Para minimizar efeito: Diminuir distância dos eletrodos Aumentar temperatura Aumentar concentração Utilizar bons condutores iônicos (ácidos e bases)
 observa-se uma aumento na voltagem e na potência de entrada. Para minimizar efeito: Diminuir distância dos eletrodos. Aumentar temperatura. Aumentar concentração. Utilizar bons condutores iônicos (ácidos e bases)")
36
Polarização por transporte de massa
Se o reagente estiver em baixa concentração e/ou a corrente elevada tende a um esgotamento de reagente na superfície do eletrodo. Velocidade da reação determinada pela velocidade de chegada de espécies reagentes na superfície do eletrodo. polarização por transporte de massa Considerando que o sistema não apresente polarização por queda ohmica e também não apresente polarização por ativação Chegada do reagente na superfície do eletrodo limita a reação

37
Espécie deve difundir até a superfície do eletrodo.
Difusão: Movimento dos íons ou espécies neutras devido à diferença de potencial químico ou concentração Sistemas eletroquímicos pode aparecer a difusão devido ao consumo de reagentes na interface eletrodo/solução Perfil de concentração:

38
Além da difusão pode ocorrer:
Convecção: Movimento de íons ou espécies neutras devido agitação Migração: Movimento de íons ou espécies neutras devido a presença de campos elétricos. Ao chegar na superfície do eletrodo a espécie reagente é consumida etapa determinante da reação é a chegada da espécie. Nesta condição – corrente elétrica é limitada pelo fluxo de espécies que chegam ao eletrodo. Como as espécies chegam ao eletrodo?

39
Depende da geometria do eletrodo
Eletrodos planos

40
Eletrodos esféricos

42
Como a corrente que circula no sistema é limitada pelo fluxo de espécies (J) que chegam à superfície do eletrodo, aplica-se Lei de Faraday I = - nFJ

43
J = mols de espécies que chegam ao eletrodo
I = - nFJ I = corrente n = número de elétrons F = Faraday J = mols de espécies que chegam ao eletrodo Isolando J j= -i/nF substitui na lei de Fick J = -D δc/δx -i/nF = -D δc/δx I = n F D δc/δx 1ª equação fundamental de cinética de difusão

44
Considerando que ocorra um salto de potencial de onde não há reação eletroquímica para onda a reação é controlada por difusão Condições iniciais e de contorno t = Cs = C T>= lim (xinf) C = Cinf T>0 x= Co=0
 C = Cinf. T>0. x=0 Co=0.")
45
Estado Transiente δC/δt = D (δ2C / δx2)
Perfil de concentração varia com o tempo usa-se a segunda lei de Fick δC/δt = D (δ2C / δx2) Segunda equação fundamental de difusão
 Segunda equação fundamental de difusão.")
46
Estado Estacionário δC/δt = D (δ2C / δx2)
δ = camada de difusão de Nernst Não á variação no perfil de concentração com o tempo: Então δC/δt = 0 δC/δt = D (δ2C / δx2) Segunda derivada de uma função = 0 Primeira derivada = constante δC/δx = const Integral resulta na equação da reta y = ax + b x = distancia do eletrodo b = concentração na superfície a = concentração zero
 Segunda derivada de uma função = 0. Primeira derivada = constante δC/δx = const. Integral resulta na equação da reta. y = ax + b. x = distancia do eletrodo. b = concentração na superfície. a = concentração. zero.")
47
Assim: δC/δx = Cinf – Csup / δ e como C sup = 0 Da 1ª equação fundamental: I = n F D δc/δx I = n F D [Cinf – Csup / δ ] i = n F D [Cinf – Csup / δ ] IL = n F D [Cinf / δ ] iL = corrente limite difusional Perfil de concentração No equilíbrio (E=0) i =0 Quando E inf i iL
![Assim: δC/δx = Cinf – Csup / δ e como C sup = 0 Da 1ª equação fundamental: I = n F D δc/δx I = n F D [Cinf – Csup / δ ] i = n F D [Cinf – Csup / δ ] IL = n F D [Cinf / δ ] iL = corrente limite difusional](http://slideplayer.com.br/slide/3626333/11/images/47/Assim%3A+%CE%B4C%2F%CE%B4x+%3D+Cinf+%E2%80%93+Csup+%2F+%CE%B4+e+como+C+sup+%3D+0+Da+1%C2%AA+equa%C3%A7%C3%A3o+fundamental%3A+I+%3D+n+F+D+%CE%B4c%2F%CE%B4x+I+%3D+n+F+D+%5BCinf+%E2%80%93+Csup+%2F+%CE%B4+%5D+i+%3D+n+F+D+%5BCinf+%E2%80%93+Csup+%2F+%CE%B4+%5D+IL+%3D+n+F+D+%5BCinf+%2F+%CE%B4+%5D+iL+%3D+corrente+limite+difusional.jpg "Perfil de concentração. No equilíbrio (E=0) i =0. Quando E inf i iL.")
48
Eletrodo rotatório mantém constante a espessura da camada de difusão
Fluxo convectivo lamelar Na camada δ não há movimento da solução – somente difusão Para cada velocidade angular o sistema atinge um estado estacionário. Vantagem- δ pode ser calculado pelas leis da hidrodinâmica δ = D 1/3 ν1/6 / 0,62 ω1/2 substituindo em: i = nFDo( δCo - Cs/ δ) chega a: ν = viscosidade cinemática da solução i = 0,62 nF D 2/3 ν-1/6 ω1/2 (Co – Cs) id = 0,62 nF D 2/3 ν-1/6 ω1/2 Co I α ω1/2
 chega a: ν = viscosidade cinemática da solução. i = 0,62 nF D 2/3 ν-1/6 ω1/2 (Co – Cs) id = 0,62 nF D 2/3 ν-1/6 ω1/2 Co. I α ω1/2.")
49
Usando as leis da hidrodinâmica, pode-se calcular δ
D = Coeficiente de difusão ω = velocidade angular ν = viscosidade cinemática da solução Como: i = n F D [Cinf – Csup / δ ] ou IL = n F D [Cinf / δ ] Subst δ Temos: i = 0,62 n F D2/3 ν-1/6 ω1/2[Cinf – Csup ] iL = 0,62 n F D2/3 ν-1/6 ω1/2[Cinf] iL vs ω1/2 linear – Levich IL diretamente proporcional a área do eletrodo e é totalmente acessível Potencial 0,0 0,4 0,8 1,2 1,6 Corrente A B C D E

50
E E D D Corrente C C Corrente B B A A Potencial Concentração 0,0 0,4
0,8 1,2 1,6 Corrente A B C D E 2 4 6 8 10 Concentração A B C D E Corrente

Apresentações semelhantes








.>")











