Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Bioinformática Alexandre L. Martins Valério A. Balani

2
Introdução A cinqüenta anos atrás, o estudo da célula se baseava fundamentalmente na observação de suas estruturas com o uso do microscópio. Bem pouco se conhecia do complexo mecanismo que se processa em nível molecular, coordenando, por meio da atividade enzimática, todo o vasto funcionamento íntimo da célula.

3
Introdução O DNA foi, há 50 anos atrás, a última grande revolucionária descoberta científica da humanidade, abrindo novos caminhos para o desenvolvimento das ciências da vida e para o nascimento de áreas multidisciplinares de estudo e pesquisa antes desconhecidas. Biologia Molecular Bioinformática Genômica Proteômica Engenharia Genética, ...

4
O que é Bioinformática? "A bioinformática é uma nova disciplina científica com raízes nas ciências da computação, na estatística e na biologia molecular. A bioinformática desenvolveu-se para enfrentar os resultados das iniciativas de seqüenciamento de genes, que produzem uma quantidade cada vez maior de dados sobre proteínas, DNA e RNA. Desse modo, os biólogos moleculares passaram a utilizar métodos estatísticos capazes de analisar grandes quantidades de dados biológicos, a predizer funções dos genes e a demonstrar relações entre genes e proteínas". Universidade de Wageningen, Holanda

5
O que é Bioinformática? Dentre as características da Bioinformática, pode-se citar: O recebimento das seqüências O tratamento de seqüências e a montagem do genoma A anotação do genoma. Base para novas hipóteses

6
O que é Bioinformática? Bioinformática: Um ramo da Biologia Computacional que se vale de “informações” para entender a Biologia. Para tanto, ela constroi ferramentas computacionais com base em “Algoritmos” que representam o comportamento dos dados biológicos, sendo este comportamento definido pela Ciência da Computação como “Estrutura de Dados”.

7
O que é Bioinformática? As características funcionais da Bioinformática são: Representação, Armazenamento e Distribuição de dados Biológicos.

8
Qual a importância da BI para os biólogos?
O que é Bioinformática? Qual a importância da BI para os biólogos? Melhor planejamento experimental, Redução de custos em P&D (homem/hora), Melhor compartilhamento de informações e Melhor Armazenamento de Informações.
, Melhor compartilhamento de informações e. Melhor Armazenamento de Informações.")
9
O que é Bioinformática? Importância
Genoma Humano: previsto para ser desenvolvido e concluído em 15 anos, foi antecipado, em cerca de 5 anos. Hoje, um novo gene, com 12 mil bases tem sua seqüência decifrada em 1 minuto, há 3 anos atrás a mesma tarefa levaria 20 minutos.

10
Breve Introdução aos Conceitos Computacionais da Bioinformática

11
Menor parte da informação que não possui um significado em si.
Dado Menor parte da informação que não possui um significado em si. Exemplo:

12
Estrutura de Dados Comportamento dos dados e suas características cuja determinação permite definir qual o melhor tipo de tratamento a eles se deve aplicar visando a obtenção de informação sobre estes.

13
Ex: 12 anos, 12 anos, 25 anos : a média dos tempos em ano é 16,3 anos.
Informação Conjunto de dados organizado de maneira a possuirem um significado que descreva um objeto. Ex: 12 anos, 12 anos, 25 anos : a média dos tempos em ano é 16,3 anos.

14
Algoritmo Processo de cálculo em que um certo número de regras formais resolvem de forma precisa ou aproximada, na generalidade, sem exceções e de forma finita, problemas da mesma natureza.

15
Exemplo de Algoritmo 1. Iniciar 2. Armazene X 3. Armazene Y
4. Some X + Y 5. Apresente o resultado 6. Finalizar

16
Linguagem Conjunto de regras gramaticais que definem a estrutura de comunicação entre o usuário e o Sistema Computacional.

17
int main() { int *i; new(i); while (i != null) new(i); }
Um Vírus em C int main() { int *i; new(i); while (i != null) new(i); }
 { int *i; new(i); while (i != null) new(i); }")
18
Sistema Computacional
Infra-estrutura na qual são feitas as implementações dos conceitos computacionais, pode ser dividido em dois conjuntos: Hardware e Software.

19
Infra-Estrutura específica para a BI

20
Sistema Operacional(SO)
Software responsável pelo gerenciamento das atividades de um sistema computacional.

21
UNIX e GNU/LINUX Estes dois sistemas fazem parte da família X (seu criador não foi o Prof. Xavier) e têm por caracterísiticas: Confiabilidade, Multiplataforma (baixa ou alta), Multiusuário, Multitarefa, Enorme gama de comandos, Não é um sistema amigavel para iniciantes, Possui um conjunto pderoso de aplicativos, POSIX (Portable Operating System Interface), Comunidade de desenvolvedores, Escrito todo em C e Baseado em arquivos texto (.txt)
, Multiusuário, Multitarefa, Enorme gama de comandos, Não é um sistema amigavel para iniciantes, Possui um conjunto pderoso de aplicativos, POSIX (Portable Operating System Interface), Comunidade de desenvolvedores, Escrito todo em C e. Baseado em arquivos texto (.txt)")
22
Por que X? O motivo da BI usar o Unix/Linux como SO preferêncial está no fato desse SO ter sido criado para desenvovimentos de software de alto desempenho em situações críticas, particularmente aquelas nas quais estão envovidas enormes quantidades de dados. A possibilidade de se usar um SO de alta performance em baixa platamorfa (Linux e FreeBSD) ou se valer dos Clusters que são construidos com base em Linux e fazem as vezes dos Supercomputadores. O fator “preço” também é importante, é possível usar sistemas X sem a necessidade de se pagar direitos autorais. Some a isso o perfil acadêmico destes softwares que já são amplamente usados em outras áreas como física e matemática.
 ou se valer dos Clusters que são construidos com base em Linux e fazem as vezes dos Supercomputadores. O fator preço também é importante, é possível usar sistemas X sem a necessidade de se pagar direitos autorais. Some a isso o perfil acadêmico destes softwares que já são amplamente usados em outras áreas como física e matemática.")
23
Linguagem de Programação Perl e BI.
A Linguagem Prática de Extração e Geração de Relatórios - The Practical Extraction and Report Language (ou Pathologically Eclectic Rubbish Lister) é uma linguagem de programação estável e multiplataforma, usada em aplicações de missão crítica em todos os setores, e é bastante usada para desenvolver aplicações web de todos os tipos, foi criada por Larry Wall em dezembro de A origem do Perl remonta ao shell scripting, Awk e à linguagem C, e está disponível para praticamente todos os sistemas operacionais, mas é usado mais comumente em sistemas Unix e compatíveis. Perl é uma das linguagens preferidas por administradores de sistema e autores de aplicações para a web. É especialmente versátil no processamento de cadeias (strings), manipulação de texto e no pattern matching implementado através de expressões regulares, além de permitir tempos de desenvolvimento curtos. A linguagem Perl já foi portada para mais de 100 diferentes plataformas, e é bastante usada em desenvolvimento web, finanças e bioinformática
 é uma linguagem de programação estável e multiplataforma, usada em aplicações de missão crítica em todos os setores, e é bastante usada para desenvolver aplicações web de todos os tipos, foi criada por Larry Wall em dezembro de A origem do Perl remonta ao shell scripting, Awk e à linguagem C, e está disponível para praticamente todos os sistemas operacionais, mas é usado mais comumente em sistemas Unix e compatíveis. Perl é uma das linguagens preferidas por administradores de sistema e autores de aplicações para a web. É especialmente versátil no processamento de cadeias (strings), manipulação de texto e no pattern matching implementado através de expressões regulares, além de permitir tempos de desenvolvimento curtos. A linguagem Perl já foi portada para mais de 100 diferentes plataformas, e é bastante usada em desenvolvimento web, finanças e bioinformática.")
24
Características da Linguagem Perl
Perl tira as melhores características de linguagens como C, awk, sed, sh, e BASIC, entre outras. Sua interface de integração com base de dados (DBI) suporta muitos bancos de dados, incluindo Oracle, Sybase, PostgreSQL, MySQL e outros. Perl tem módulos para trabalhar com HTML, XML, e outras linguagens de markup. Perl suporta Unicode.Perl permite programação procedural e orientada a objetos. Perl pode acessar bibliotecas externas em C/C++ através de XS ou SWIG. Perl é extensível. Existem milhares de módulos disponíveis no Comprehensive Perl Archive Network (CPAN). O interpretador Perl pode ser embutido em outros sistemas.
 suporta muitos bancos de dados, incluindo Oracle, Sybase, PostgreSQL, MySQL e outros. Perl tem módulos para trabalhar com HTML, XML, e outras linguagens de markup. Perl suporta Unicode.Perl permite programação procedural e orientada a objetos. Perl pode acessar bibliotecas externas em C/C++ através de XS ou SWIG. Perl é extensível. Existem milhares de módulos disponíveis no Comprehensive Perl Archive Network (CPAN). O interpretador Perl pode ser embutido em outros sistemas.")
25
Exemplo de código em Perl
printf “Oi mundo!! \n”; Em C void main() { printf (“Oi mundo!! \n”); }
 { printf ( Oi mundo!! \n ); }")
26
Banco de Dados Bancos de dados, (ou bases de dados), são conjuntos de dados com uma estrutura regular que organizam informação. Um banco de dados normalmente agrupa informações utilizadas para um mesmo fim. Um banco de dados é usualmente mantido e acessado por meio de um software conhecido como Sistema Gerenciador de Banco de Dados (SGBD). Normalmente um SGBD adota um modelo de dados, de forma pura, reduzida ou extendida. Muitas vezes o termo banco de dados é usado como sinônimo de SGDB. O modelo de dados mais adotado hoje em dia ó o modelo relacional, onde as estruturas têm a forma de tabelas, compostas por linhas e colunas.
, são conjuntos de dados com uma estrutura regular que organizam informação. Um banco de dados normalmente agrupa informações utilizadas para um mesmo fim. Um banco de dados é usualmente mantido e acessado por meio de um software conhecido como Sistema Gerenciador de Banco de Dados (SGBD). Normalmente um SGBD adota um modelo de dados, de forma pura, reduzida ou extendida. Muitas vezes o termo banco de dados é usado como sinônimo de SGDB. O modelo de dados mais adotado hoje em dia ó o modelo relacional, onde as estruturas têm a forma de tabelas, compostas por linhas e colunas.")
27
Bancos de Dados Sistema de Gerenciamento de Banco de Dados MySQL
Construção Manipulação Administração MySQL PostgreSQL Oracle sqlServer MySQL Gratuíto Código Aberto Acesso Veloz aos Dados

28
Bancos de Dados

29
Bancos de Dados Fonte: GOLD[TM] Genomes OnLine Database
![Bancos de Dados Fonte: GOLD[TM] Genomes OnLine Database](http://slideplayer.com.br/slide/376138/2/images/29/Bancos+de+Dados+Fonte%3A+GOLD%5BTM%5D+Genomes+OnLine+Database.jpg)
30
INSDC – International Nucleotide Sequence Database Colaboration
Bancos de Dados Primários: GenBank EBI-EMBL (European Bioinformatics Institut) DDBJ (DNA Data Bank of Japan) PDB (Protein Data Bank) Secundários: PIR (Protein Information Resource) SWISS-PROT INSDC – International Nucleotide Sequence Database Colaboration
 DDBJ (DNA Data Bank of Japan) PDB (Protein Data Bank) Secundários: PIR (Protein Information Resource) SWISS-PROT. INSDC – International Nucleotide Sequence Database Colaboration.")
31
Nucleic Acids Research
Bancos de Dados Funcionais: KEGG (Kyoto Encyclopedia of Genes and Genomes) Mapas metabólicos de organismos com genoma completamente ou parcialmente seqüenciados Estruturais: Mantém dados sobre estrutura de proteínas Nucleic Acids Research
 Mapas metabólicos de organismos com genoma completamente ou parcialmente seqüenciados. Estruturais: Mantém dados sobre estrutura de proteínas. Nucleic Acids Research.")
32
BD de Seqüências EMBL (http://www.ebi.ac.uk/embl)
Há uma quantidade gigantesca de informação sobre biomoléculas em BD públicos Mais de 348 BD BD de seqüências de nucleotídeos EMBL ( GenBank ( DDBJ ( UniGene ( BD de seqüências de proteínas SWISS-PROT, TrEMBL ( PIR ( BD de motivos Pfam ( PROSITE ( BD de estruturas macromoleculares 3D PDB (

33
Usos de BD de Seqüências
O que se pode descobrir sobre um gene por meio de uma busca a um BD? Informação evolutiva: genes homólogos, freqüências dos alelos, ... Informação genômica: localização no cromossomo, introns, ORFs, regiões reguladoras, ... Informação estrutural: estruturas da proteína correspondente, tipos de folds, domínios estruturais, ... Informação de expressão: expressão específica a um dado tecido, fenótipos, doenças, ... Informação funcional: função molecular/enzimática, papel em diferentes rotas, papel em doenças, ...

34
Busca em BD de Seqüências
O que queremos saber sobre a seqüência? Ela é similar ao algum gene conhecido? Quão próximo é o melhor match? Significância? O que sabemos sobre este gene? Genômica (localização no cromossomo, regiões reguladoras, ...) Estrutural (estrutura conhecida? ...) Funcional (molecular, celular e doença) Informação evolutiva Este gene é encontrado em outros organismos? Qual é sua árvore taxonômica?
 Estrutural (estrutura conhecida ...) Funcional (molecular, celular e doença) Informação evolutiva. Este gene é encontrado em outros organismos Qual é sua árvore taxonômica")
35
NCBI e Entrez A mais usada interface para a recuperação de informação de BD biológicos é o sistema Entrez do NCBI ( NCBI (National Center for Biotechnology Information) O sistema Entrez tira vantagem do fato que há relacionamentos lógicos pré-existentes entre as entradas indíviduas encontradas em diversos BD públicos Por um exemplo, um artigo no PuBMed pode descrever o sequenciamento de um gene cuja seqüência aparece no GenBank A seqüência de nucleotídeos, por sua vez, pode codificar o produto de uma proteína cuja seqüência está armazenada em um BD de proteínas A estrutura 3D desta proteína pode ser conhecida - as coordenadas da estrutura podem aparecer em um BD de estruturas Finalmente, o gene pode ter sido mapeado para uma região específica do cromossomo - BD de mapeamento A existência dessas conexões naturais, levou ao desenvolvimento de um método por meio do qual toda a informação poderia ser encontrada sem ter que visitar sequencialmente BD distintos
 O sistema Entrez tira vantagem do fato que há relacionamentos lógicos pré-existentes entre as entradas indíviduas encontradas em diversos BD públicos. Por um exemplo, um artigo no PuBMed pode descrever o sequenciamento de um gene cuja seqüência aparece no GenBank. A seqüência de nucleotídeos, por sua vez, pode codificar o produto de uma proteína cuja seqüência está armazenada em um BD de proteínas. A estrutura 3D desta proteína pode ser conhecida - as coordenadas da estrutura podem aparecer em um BD de estruturas. Finalmente, o gene pode ter sido mapeado para uma região específica do cromossomo - BD de mapeamento. A existência dessas conexões naturais, levou ao desenvolvimento de um método por meio do qual toda a informação poderia ser encontrada sem ter que visitar sequencialmente BD distintos.")
36
Mais que NCBI Links para anotações funcionais fora do NCBI
Gene Ontology - nomes padrões para: Funções moleculares Localização celular Processos Links para o BD KEGG (vias)
")
37
Alinhamento de Seqüências
Possibilitar ao pesquisador determinar se duas seqüências apresentam suficiente similaridade tal que uma inferência sobre homologia possa ser justificada Homologia: significa dizer que duas (ou mais) seqüências tem um ancestral comum História evolutiva Similaridade: é uma medida da qualidade do alinhamento entre duas seqüências, baseada em algum critério Não se refere a nenhum processo histórico Apenas uma comparação das seqüências com algum método É uma afirmação logicamente mais fraca
 seqüências tem um ancestral comum. História evolutiva. Similaridade: é uma medida da qualidade do alinhamento entre duas seqüências, baseada em algum critério. Não se refere a nenhum processo histórico. Apenas uma comparação das seqüências com algum método. É uma afirmação logicamente mais fraca.")
38
Relação entre Seqüências

39
Alinhamento de Seqüências
Programas mais utilizados: ClustalW Multialin FASTA Blast 2 sequences Blast On-line

40
Alinhamento de Seqüências
Alinhamento Global e Local

41
Alinhamento Global e Local
Seqüências são comparadas como um todo Útil quando temos seqüências que diferem pouco entre si Inclui gaps Local O alinhamento localiza fragmentos de seqüências que são mais similares Algumas vezes não inclui gaps Muitas proteínas não apresentam um padrão global de similaridade Mosaico de domínios modulares Alinhamento de seqüências de nucleotídeos de um mRNA processado (spliced) com sua seqüencia genômica (Exon/Intron)
 com sua seqüencia genômica (Exon/Intron)")
42
Alinhamento de Seqüências
Unidade pareada (match): + Espaços (gaps): - Não pareadas (mismatch): -
: + Espaços (gaps): - Não pareadas (mismatch): -")
43
Alinhamento de Seqüências

44
Alinhamento de Seqüências

45
Alinhamento de Seqüências

46
Alinhamento de Seqüências

47
Alinhamento de Seqüências

48
Alinhamento de Seqüências
Blast 2 Sequences

49
Projetos Genoma Shotgun Shotgun hierárquico

50
Bioinformática Montagem do genoma

52
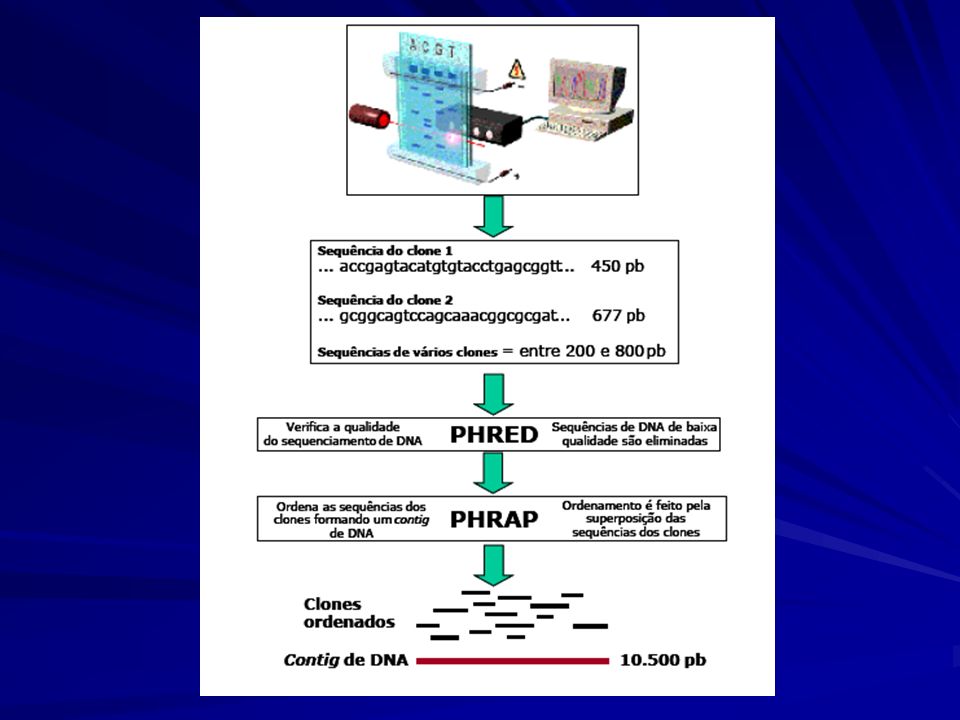
Identifica e atribui valor de qualidade para cada nucleotídeo
Base Calling Dados Brutos Programa de do Seqüênciador Base calling PHRED Identifica e atribui valor de qualidade para cada nucleotídeo

53
Base Calling PHRED Reconhece arquivos .SCF, .ABI e .MegaBACE ESD
Reconhece os dados brutos do seqüenciador Atribui valores de qualidade aos nucleotídeos Gera arquivos de saída com informações sobre o basecall e os valores de qualidade (FASTA e PHD)
")
54
Base Calling Cálculo Algorítmo – Métodos de Análise de Fourier.
Qualiadade: probabilidade de erro PHRED Quality -log (Pe) Ex: Valor 20 para uma posição nucleotídica significa uma chance em 100 de estar errada Valor 30 para uma posição nucleotídica significa uma chance em 1000 de estar errada
 Ex: Valor 20 para uma posição nucleotídica significa uma chance em 100 de estar errada. Valor 30 para uma posição nucleotídica significa uma chance em 1000 de estar errada.")
55
Phred – qualidade dos reads
Alta qualidade Média qualidade Baixa qualidade

56
Mascaramento de Vetores
Retirada de seqüências contaminantes: Partes de vetores de clonagens DNA adaptores Programa mais utilizado é o Cross_match

57
Mascaramento de Vetores

58
Agrupamento de Seqüências
Software de montagem (Assembler) PHRAP CAP3 TIGR Assembler PHRAP Phragment Assembly Program Leitura do base call Montagem dos contigs
 PHRAP. CAP3. TIGR Assembler. PHRAP. Phragment Assembly Program. Leitura do base call. Montagem dos contigs.")
59
Agrupamento de Seqüências
Pontos Chaves Uso de seqüências com alta qualidade Uso de informações de qualidade computadas internamente e fornecidas pelo usuário Informações sobre as montagens realizadas Projetos Genoma = contíguo genômico Projetos Transcriptoma = seqüências dos genes expressos

60
O Phrap Assembler (monta as sequencias contíguas usando as reads).
Contig 1 reads Região de sobreposição Contig 2

61
Visualização e Montagem
Progamas Phrapview ou Consed

62
Standen Package Pregap4

63
Standen Package Gap4

64
Standen Package Gap4

65
Standen Package Spin

66
Standen Package Trev

67
Fluxo de dados Sequenciador Phred Phd2fasta Cross_match Consed Phrap
Indireto Cross_match Consed Phrap

68
Análise de Genomas Então, o que fazer com um genoma completo?
Afinal, um genoma seqüenciado consiste apenas de um infinidade de bases em uma ordem definida Análise é obviamente necessária a fim de se obter informações biologicamente interessantes. A análise de um genoma cobre muitos aspectos diferentes

69
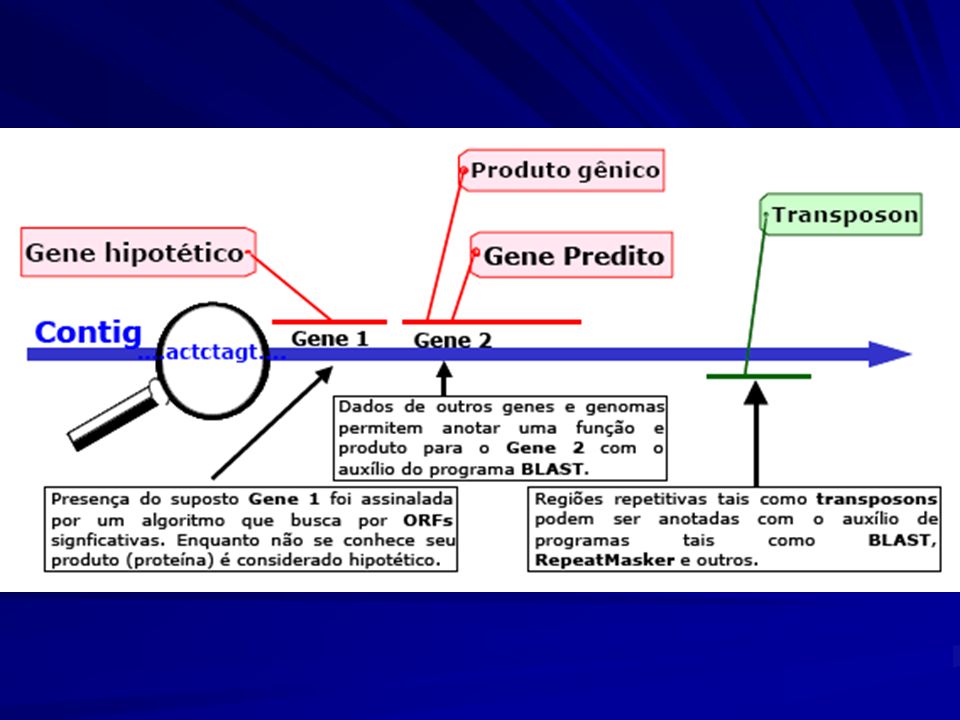
Anotação Gênica RepeatMasker Genscan tRNAscan-SE BLAST
InterproGeneOntology GenomeScan

70
Definição da localização dos genes (regiões codificadoras, regiões reguladoras)
Predição de genes ab initio usando software baseado em regras e padrões. Identificação de genes por meio de alinhamento com proteínas conhecidas e seqüências EST Predição de genes por meio de similaridade com proteínas e seqüências EST em outros organismos Predição de genes por meio de comparação com outros genomas Regiões conservadas são provavelmente regiões codificadoras ou reguladoras

72
Anotação Gênica Algorítimo gene-finder chamado BGF (BGI GeneFinder) baseado no GenScan e FgeneSH Teste com Drosophila Predição: genes Oficial: genes

73
E a Bioinformática não pára por ai...

Apresentações semelhantes