Carregar apresentação
A apresentação está carregando. Por favor, espere

1
DOENÇA DE FABRY Natalia Fabris Gama 20071130055
Universidade Federal do Estado do Rio de Janeiro – UNIRIO Escola de Medicina e Cirurgia – EMC Disciplina de Genética II DOENÇA DE FABRY Natalia Fabris Gama

2
HISTÓRICO

3
Histórico Abril, 1897 Johannes Fabry (em paciente de 13 anos)
Dezembro, 1897 William Anderson (em paciente de 39 anos) Johannes Fabry William Anderson
 Johannes Fabry. William Anderson.")
4
Histórico 1898 Descrição da doença pelos dois dermatologistas
1900 Os dermatologistas concluíram que os pacientes que sofriam de “angioqueratoma difuso” e tinham uma expectativa de vida reduzida Por isso, embora mais comumente chamada de doença de Fabry (DF), também é conhecida como doença de Anderson-Fabry
, também é conhecida como doença de Anderson-Fabry.")
5
Definição A DF é uma doença hereditária de depósito lisossômico
Erro inato do metabolismo dos glicoesfingolipídeos Rara, crônica, progressiva, sistêmica e grave

6
GENÉTICA

7
Genética Herança ligada ao X Gene afetado é o GALA região Xq22
Defeito genético que produz a doença é extremamente heterogêneo “Mutações privadas” – só de determinada família

8
Genética Homens hemizigotos – alta penetrância
Mulheres heterozigotas – expressividade variável Não são simples portadoras Alterações sistêmicas relacionadas à doença Aumento da morbidade e mortalidade Ligada ao X recessiva (?)
")
9
ETIOLOGIA

10
Etiologia Mutações do gene que codifica a enzima lisossômica α-galactosidase A (α-GAL) Acúmulo progressivo de glicoesfingolipídeos neutros com resíduos terminais α-galactosil (principalmente de globotriasilceramida ou GL-3) no plasma e no lisossomo de várias células células endoteliais, pericitos, células musculares lisas dos vasos sanguíneos, miócitos cardíacos, células epiteliais dos glomérulos e túbulos renais, células ganglionares do sistema nervoso autônomo, células da córnea e células do tecido conectivo.
 no plasma e no lisossomo de várias células. células endoteliais, pericitos, células musculares lisas dos vasos sanguíneos, miócitos cardíacos, células epiteliais dos glomérulos e túbulos renais, células ganglionares do sistema nervoso autônomo, células da córnea e células do tecido conectivo.")
11
Etiologia Em células endoteliais, pericitos e células musculares lisas sanguíneas protuberâncias no lúmen dos vasos estreitamentos e dilatações isquemia e infarto As áreas mais afetadas pela oclusão de pequenos vasos sangüíneos são rins, coração, sistema nervoso e pele Outros tipos celulares aumento do tamanho celular organomegalias e disfunção visceral

12
EPIDEMIOLOGIA

13
Epidemiologia Todas as raças , mas mais frequente na raça branca
1: homens 1: nascidos vivos No Brasil 220 pacientes No mundo mais de 25 mil pessoas atingidas com a DF Segunda alteração mais frequente por acúmulo lisossômico nos humanos Incidência subestimada variantes intermediárias post mortem

14
Epidemiologia

15
MANIFESTAÇÕES CLÍNICAS

16
Clínica Geralmente decorrem-se 12 anos entre o início dos sintomas e o estabelecimento do diagnóstico Nas mulheres, o aparecimento dos sintomas ocorre, em média, 6 anos mais tarde que nos homens Estima-se que 40% dos pacientes são tratados como portadores de “reumatismo” As manifestações iniciais mais freqüentes são geralmente dermatológicas, neurológicas e gastrointestinais

17
Clínica A dor é um sintoma precoce, muitas vezes debilitante, observada em cerca de 77% dos pacientes Mãos e pés Crises de dores torturantes – dor ardente acompanhada de formigamento Pode ser intermitente ou contínua “Crise de Fabry” Dor aguda que dura desde minutos até dias, e que podem acompanhar-se de fadiga, febre de baixo grau e artralgia Mais de 50% do pacientes apresentam dores abdominais, distensão, crises alternadas de constipação e diarréia, falta de apetite, saciedade precoce, náuseas e vômitos

18
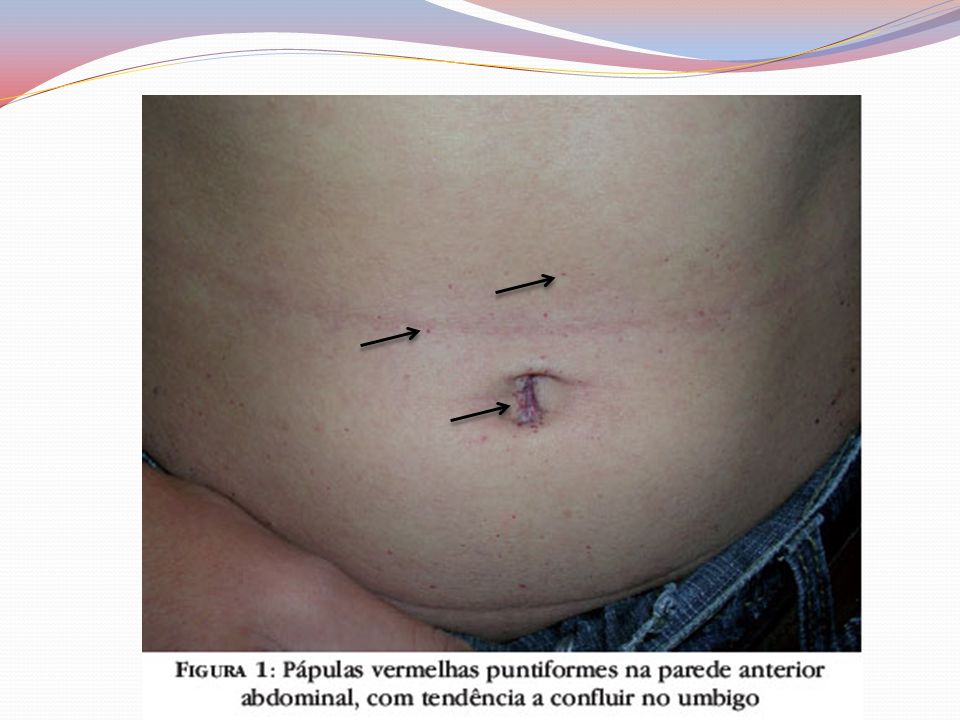
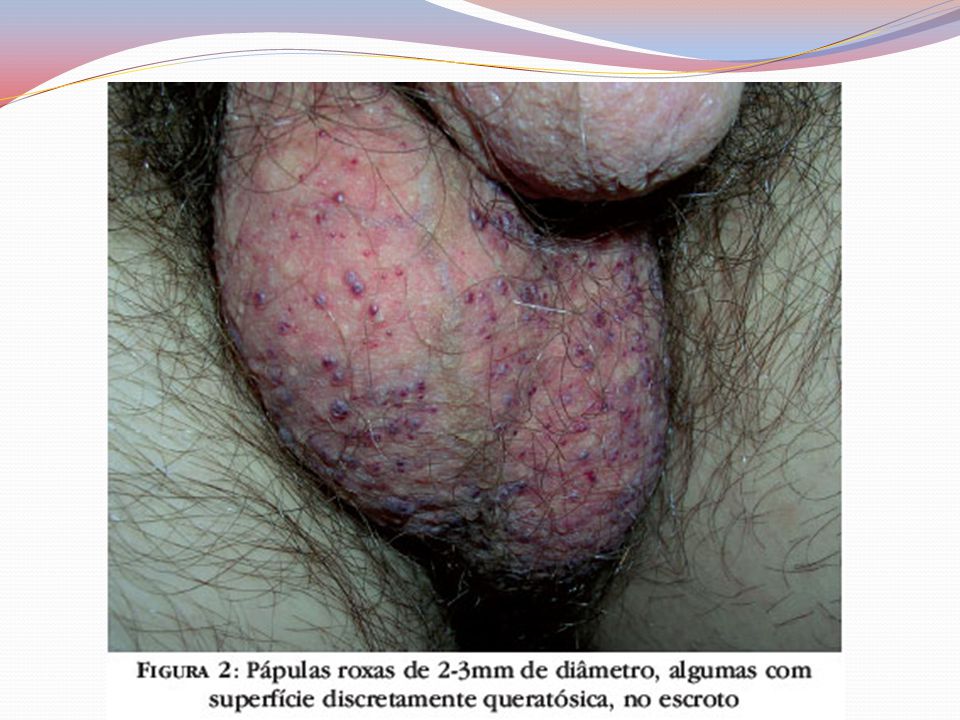
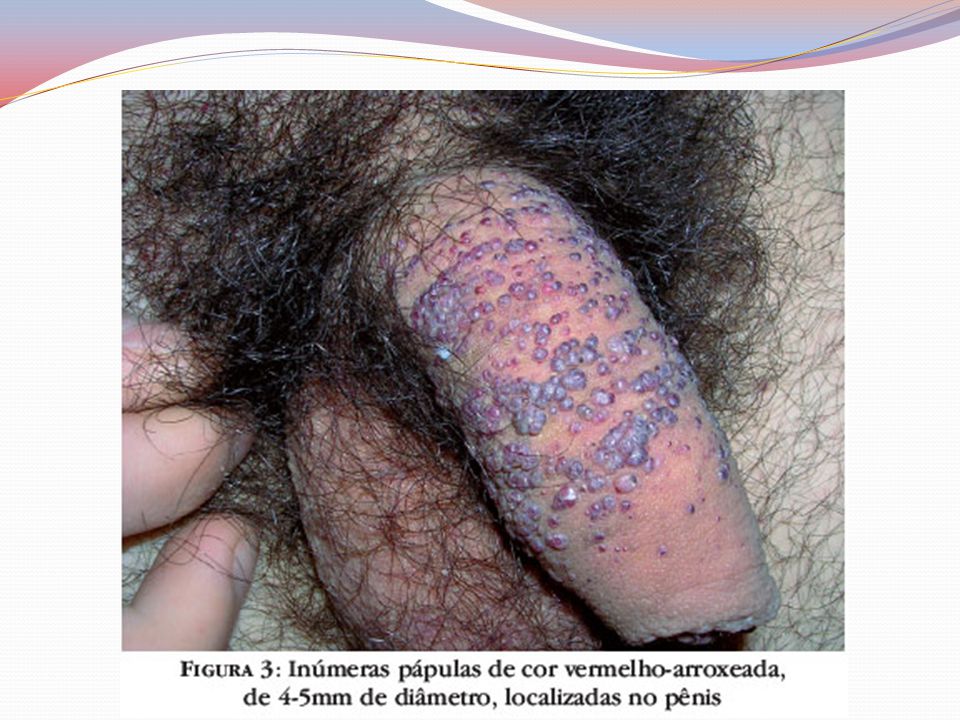
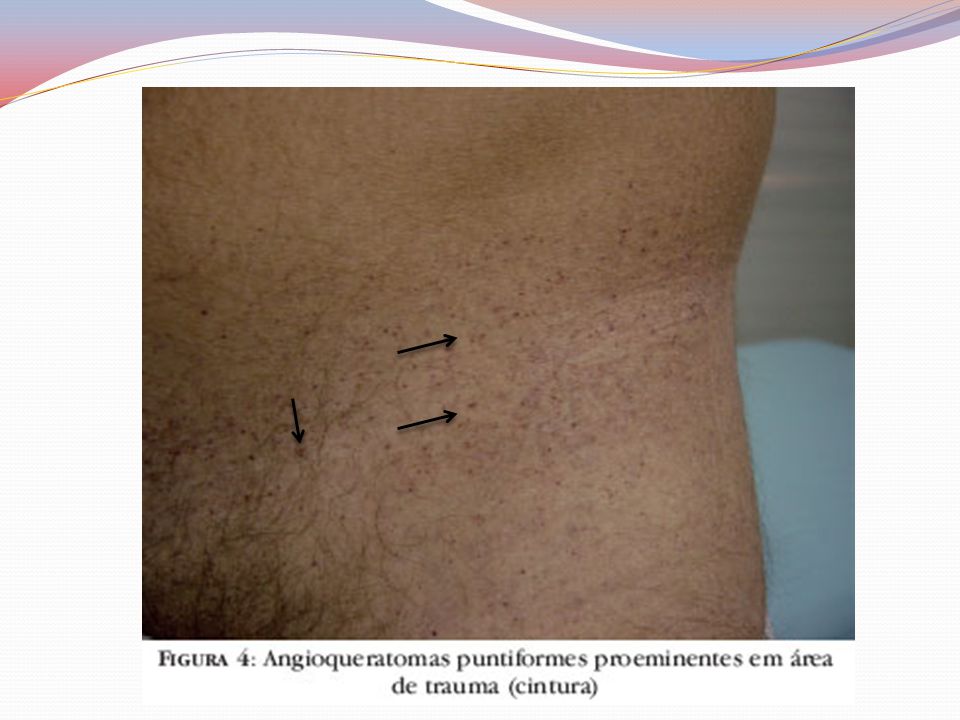
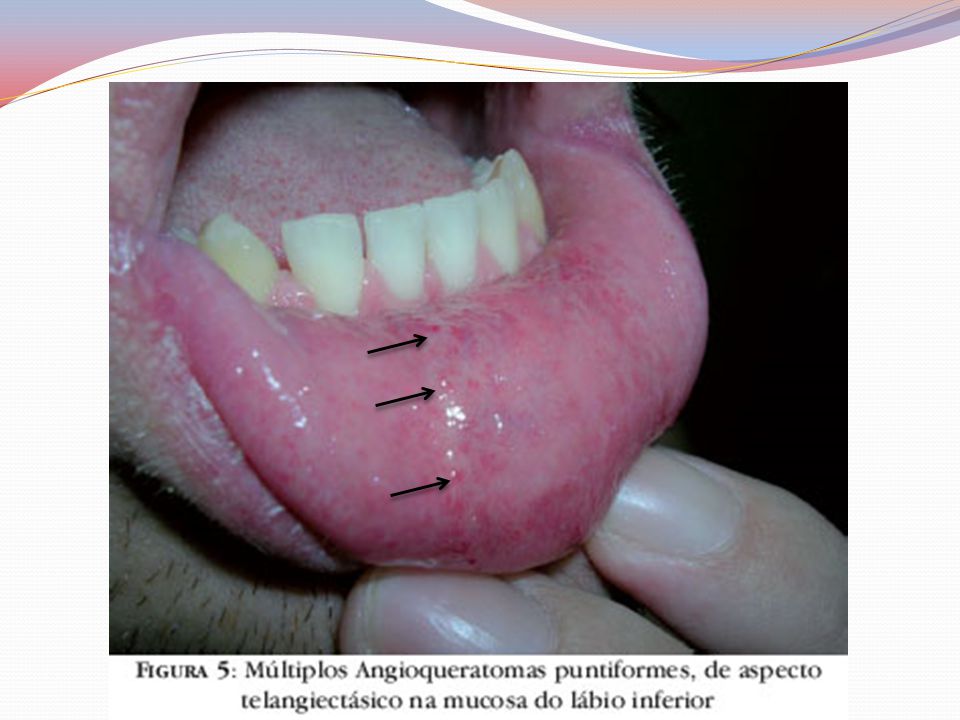
Clínica Angioqueratomas cutâneos disseminados
Pápulas eritematovinhosas ou azul-enegrecidas, com até 10mm, não se alteram à vitriopressão Dispostos em grupo e de forma simétricas “Roupa de banho” periumbilical às coxas Acometimento de mucosas – aspecto telangiectásicos

24
Clínica Hipoidrose ou anidrose com pele seca e intolerância ao calor e ao exercício Dano seletivo de nervos autônomos Depósito de GL-3 nos ácinos sudoríparos e/ou À isquemia devida ao comprometimento dos vasos sanguíneos que nutrem tanto os nervos autônomos quanto as glândulas sudoríparas Hipotricose corporal difusa Edema nas pálpebras e extremidades Lesões de córnea e cristalino

25
Córnea verticilata

26
Órgão Sinais e sintomas Pele - Angioqueratomas Sistema nervoso periférico Acroparestesias - Alterações da sudorese: • Hipohidrose • Anidrose • Hiperhidrose - raramente e em mulheres Sistema gastrointestinal - Náusea - Dores abdominais - Diarréia ou constipação em episódios alternados Visão - Córnea verticilata - Tortuosidade dos vasos - Cataratas subcapsulares

27
Órgão Sinais e Sintomas Coração (Tardio) - Hipertrofia e disfunção ventricular esquerda - Disfunção das válvulas cardíacas - Anomalias de circulação Rim - Proteinúria - Declínio progressivo na taxa de filtração glomerular com evolução para falência renal Sistema nervoso central - Ataques isquêmicos transitórios - AVE isquêmico Audição Zumbido Vertigem - Hipoacusia Qualidade de vida - Deterioração da qualidade de vida

28
Clínica Alguns doentes apresentam dismorfia facial de diferentes graus
lóbulos das orelhas proeminentes, sobrancelhas espessas, fronte deprimida, ângulo nasal pronunciado, nariz grande, ponte supraorbitária proeminente, base nasal larga e rasgos toscos Depressão, ideação suicida e demência

29
DIAGNÓSTICO

30
Diagnóstico Hemizigotos Heretozigotas
Níveis da α-GAL em lágrimas, plasma, leucócitos, cultura de fibroblastos cutâneos, em gotas de sangue seco colhidas em papel-filtro Presença de GL-3 no sedimento urinário, plasma ou cultura de fibroblastos Análise DNA CONFIMATÓRIO Níveis de α-GAL podem estar normais (inativação aleatória do X) Análise de DNA CONFIMATÓRIO
 Análise de DNA CONFIMATÓRIO.")
31
Diagnóstico Pré-natal “Teste do pezinho” extendido
Baixa atividade da α- GAL em biópsia de vilo coriônico Cultura de células do líquido amniótico, quando se tratar de fetos com cariótipo XY Em caso da mutação familiar da α-GAL ser conhecida, o estudo molecular pode suplantar ou confirmar o diagnóstico enzimático “Teste do pezinho” extendido Clínicas particulares

32
TRATAMENTO

33
Tratamento Multidisciplinar clínicos, dermatologistas, neurologistas, cardiologistas, nefrologistas e geneticistas Terapia não específica suporte Angioqueratomas: eletrocoagulação, crioterapia, exérese cirúrgica ou Laser Acroparestesias e crises de Fabry: difenilidantoína, carbamazepina, gabapentina, oxacarbazepina ou topiramato Doença vascular cerebral e retiniana: realiza- se prevenção com agentes antiplaquetários ou anticoagulantes. Doença renal: controle da hipertensão arterial, diálise, até transplante renal. Uso IECA Doença cardíaca: drogas antiarrítmicas, marcapasso até transplante cardíaco.

34
Tratamento Terapia específica
Terapia de reposição enzimática (TRE): células podem incorporar uma enzima do meio extracelular e utilizá-la para seu metabolismo normal. Administradas por via intravenosa a cada 15 dias Terapia gênica (em fase experimental): acrescentar um gene normal da α-GAL ao DNA do doente, passando este a produzir a enzima normalmente Chaperonas (em fase de estudo): realce enzimático, útil para os doentes que possuem variantes instáveis da α-GAL, retida no retículo endoplasmático, mas conservam atividade enzimática residual. Utilizam-se pequenas moléculas sintéticas que, atuando como chaperonas, resgatam a α-GAL residual transportando-a para os lisossomos. Administrada por via oral
: células podem incorporar uma enzima do meio extracelular e utilizá-la para seu metabolismo normal. Administradas por via intravenosa a cada 15 dias. Terapia gênica (em fase experimental): acrescentar um gene normal da α-GAL ao DNA do doente, passando este a produzir a enzima normalmente. Chaperonas (em fase de estudo): realce enzimático, útil para os doentes que possuem variantes instáveis da α-GAL, retida no retículo endoplasmático, mas conservam atividade enzimática residual. Utilizam-se pequenas moléculas sintéticas que, atuando como chaperonas, resgatam a α-GAL residual transportando-a para os lisossomos. Administrada por via oral.")
35
TRE Deve ser utilizada por toda a vida quantidade da enzima no plasma é rapidamente depletada precisa de infusões constantes A tolerância boa, com exceção de reações leves ou moderadas associadas à infusão, produto da formação de anticorpos IgG não neutralizantes Finalidade prevenir o desenvolvimento de doença nos pacientes jovens e evitar, quando não reverter, a progressão da disfunção orgânica nos doentes mais velhos

36
TRE Indicações Resultados Hemizigotos Heterozigotas
Menores de 16 anos: assim que houver sintomas ou sinais Maiores de 16 anos: no momento do diagnóstico Heterozigotas quando houver sintomas significativos ou acometimento de órgão nobre Resultados Diminuição da frequência das crises de dor, das alterções cardíacas e do depósito de GL-3 nos rins e na pele Melhora a sudorese, os sintomas gastrointestinais, pulmonares e a audição

37
TRE Betagalsidade Alfagalsidade Registro ANVISA
Aprovada pelo FDA e EMEA Custo anual U$215,000.00 1mg/kg/dose Não há registro ANVISA Aprovada pelo EMEA Custo anual U$215,000.00 0,2mg/kg/dose

38
ACONSELHAMENTO GENÉTICO

39
Aconselhamento genético
Todas as filhas de um hemizigoto herdarão a doença, visto que herdarão o X que tem a mutação de seu pai Nenhum dos seus filhos de um hemizigoto herdará a doença, já que eles herdarão o cromossomo Y de seu pai Metade dos filhos e metade das filhas de uma heterozigota serão acometidas, uma vez que ela tem um X com a mutação e outro sem

40
BIBLIOGRAFIA Tratado de medicina Interna, Harrison, 17ª edição, editora Mc Graw Hill, 2009 Tratado de Clínica Médica, Antônio Carlos Lopes, 1ª edição, editora Roca, 2006

41
Associação Brasileira de Pacientes Portadores da Doença de Fabry e seus Familiares

Apresentações semelhantes









, e que pode levar a incapacitação funcional.>")









>")