Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Caso Clínico Síndrome de Potter
Apresentação: Eder Ferreira Soares (interno ESCS) Geneticista: Romina S. Heredia Garcia Silva (R2) Coordenação: Paulo R. Margotto Brasília, 19 de julho de 2011 ESCS UTI Neonatal do HRAS Hospital Regional da Asa Sul
 Geneticista: Romina S. Heredia Garcia Silva (R2) Coordenação: Paulo R. Margotto. Brasília, 19 de julho de ESCS. UTI Neonatal do HRAS. Hospital Regional da Asa Sul.")
2
Dra. Romina G. Silva, Dr. Paulo R. Margoto, Do Éder F. Soares

3
Antecedentes Pré-natais
Mãe, 19 anos, internada na UGO/HRAS desde 18/03/11 devido a quadro de oligoâmnio severo; Realizou 14 dias de Amoxicilina, com término no dia 10/05/2011; Sorologias maternas: não há sorologias anotadas no prontuário, porém mãe refere ter realizado todas e que as mesmas se encontram descritas no cartão pré-natal; Esteróide pré-natal: 2 doses de betametasona (março 2011); Nega etilismo ou tabagismo; TS mãe: O+; DUM: 18/09/10; Mãe: G2P0A1; IG: 34+3 (até 02/06/11).
; Nega etilismo ou tabagismo; TS mãe: O+; DUM: 18/09/10; Mãe: G2P0A1; IG: 34+3 (até 02/06/11).")
4
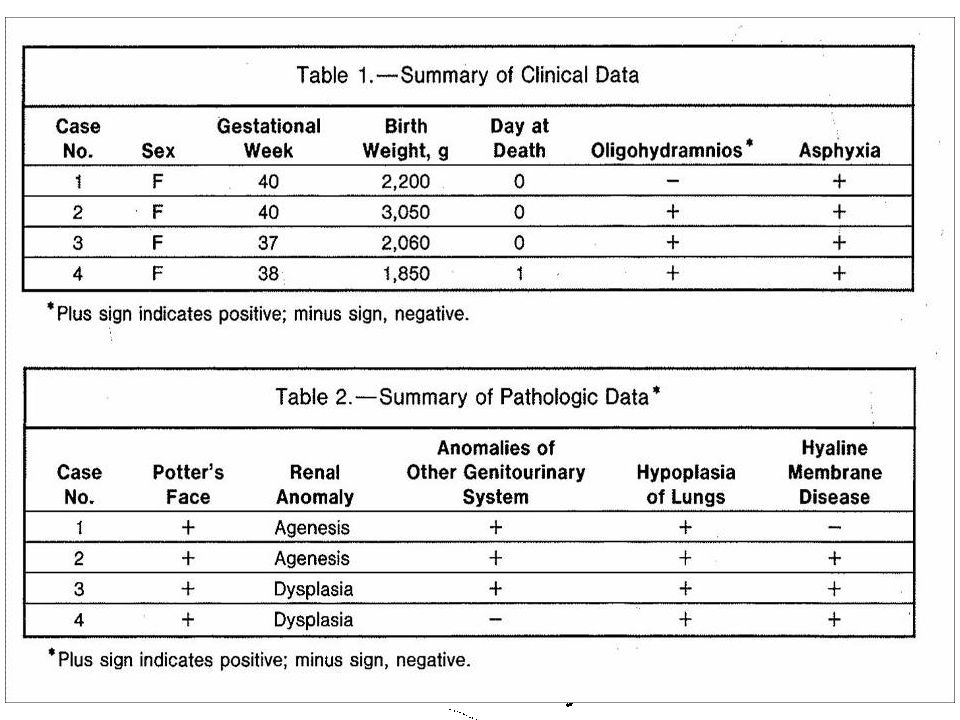
Identificação RN de RNSV, feminino; Nascimento: 02/06/2011 às 17:36h;
Parto cesáreo: ILA 0 a esclarecer + ROPREMA; Apgar: 3,5,5; Reanimação: sim, VPP sob máscara; Peso: 1680g; Est.: 38cm; PC: 29,5cm; Intercorrências na gestação: ILA zero.

5
Evolução Parto cesáreo pélvico; Ausência de líquido amniótico;
Não chorou ao nascer; Colocado em berço aquecido e secado; Aspiradas boca e narinas; Cianose central, bradicárdico, hipotenso, movimentos respiratórios irregulares;

6
Evolução Realizada VPP, sem melhora do quadro;
Feita intubação traqueal com cânula 3.0 e aumentada pressão e fluxo do CFR: sem melhora; Feito cateterismo venoso: 2 expansões com SF0,9% 10ml cada; Realizada massagem cardíaca mais 2 doses de adrenalina: persistência da bradicardia, perfusão ruim e ausência de diurese;

7
Evolução Feita nova dose de adrenalina e bicarbonato 5ml: 5ml AD. Repetido bicarbonato; Oitenta ml de volume infundidos em 1h; Melhora da perfusão e da bradicardia (FC: 110 – 125bpm), mas continuava sem diurese; Encaminhado para UTI às 18:45h.
, mas continuava sem diurese; Encaminhado para UTI às 18:45h.")
8
Evolução Exame Físico:
Cabeça: normocéfalo, fontanela anterior aberta e normotensa. Orelhas: implantação baixa (?). Boca: simétrica. AR: expansibilidade ruim; tiragem intercostal. ACV: RCR 2T, bulhas normofonéticas, sem sopro. FC: 110 – 115bpm. Pulsos impalpáveis em MMSS. Abdome: flácido, umbigo 2A e 1V. Genitália: edema de grandes e pequenos lábios. Ânus: pérvio. MMII: perfusão lentificada. Ortolani negativo. Dorso: sem lesões. Capurro: 33+6. Índice ponderal: 1680/54872 = 3.
. Boca: simétrica. AR: expansibilidade ruim; tiragem intercostal. ACV: RCR 2T, bulhas normofonéticas, sem sopro. FC: 110 – 115bpm. Pulsos impalpáveis em MMSS. Abdome: flácido, umbigo 2A e 1V. Genitália: edema de grandes e pequenos lábios. Ânus: pérvio. MMII: perfusão lentificada. Ortolani negativo. Dorso: sem lesões. Capurro: Índice ponderal: 1680/54872 = 3.")
9
Cortesia Dr. Paulo R. Margotto

10
Cortesia Dr. Paulo R. Margotto

11
Cortesia Dr. Paulo R. Margotto

12
Evolução Hipóteses diagnósticas: Conduta: RNP/PIG simétrico
Asfixia perinatal Hipoplasia pulmonar ? Sindrômico ? Sepse ? Membrana hialina ? Conduta: Encaminhado a UTI; Neo em incubadora aquecida; Intubado com cânula 3; Cateterismo umbilical.

13
Evolução UTI: Soro fisiológico (20ml/Kg em 30’);
HV com TIG 5 + dopamina 5 + dobutamina 10;

14
Evolução Às 20:00h: Ventilação mecânica com PI 40x6, FiO² 100%, FR 60ipm; Sem relato de diurese até o momento; Dreno tórax: secreção sanguinolenta em moderada quantidade; SOG: sem secreção; EF: RN GRAVÍSSIMO, chocado, desidratado; Pouca expansibilidade torácica; Bulhas abafadas. FC: 96 – 100bpm; Estertores bolhosos em HTE. PSaO² (saturação de O2):35 – 45%; Fígado a 2cm do RCD.
:35 – 45%; Fígado a 2cm do RCD.")
15
Evolução Conduta: Dopa 7,5 e dobuta 10;
Solicitado concentrado de hemácias + plasma; Nova expansão com SF 0,9% (15ml/Kg); Adrenalina 0,3mg; Resgatar Rx tórax pós-drenagem torácica; Fentanil + Cefepime + Amicacina.
; Adrenalina 0,3mg; Resgatar Rx tórax pós-drenagem torácica; Fentanil + Cefepime + Amicacina.")
16
Evolução Às 20:25h: Rx tórax: TOT pouco alto; dreno com extremidade em cavidade abdominal (?). CD: dopamina 10 e dobutamina 20; nova dose de adrenalina. Às 21:30h: Dissecção de veia jugular interna esquerda e reposicionamento dreno; Mantida bradicardia e PSaO² de 25 – 30%; CD: nova etapa SF0,9% (15ml/Kg); adrenalina contínua (0,5ug/g/min).
; adrenalina contínua (0,5ug/g/min).")
17
Evolução Às 22:15h: Melhora discreta da expansão pulmonar. PI: 50; Perfusão ruim. FC: 92bpm. PSaO² (saturação de O2): 18 – 22%; Ausência de diurese; Gasometria: pH 6,74, PCO² 150, PO² 15.9, BE -16. Às 22:50h: Dreno fora do tórax; pneumotórax refazendo-se; CD: Repassado dreno de tórax;

18
Evolução FiO² 100%, PI 50, PSaO² 21%, FC: 52bpm; Às 3:10h:
GRAVÍSSIMO estado geral; FC: 85 – 90bpm, RCR 3T, B3, bulhas abafadas; Ausência de diurese; CD: solicitados ecografias cardíaca, cerebral e abdominal. 13h de vida: FiO² 100%, PI 50, PSaO² 21%, FC: 52bpm; Palidez importante, péssima perfusão, cianose; Diurese ausente; Ecografia abdominal: agenesia renal bilateral. Ecografia transfontanelar: cisto plexo direito.

19
Evolução Às 9:30h: Às 11:05: Permanece em estado GRAVÍSSIMO.
Constatado óbito.

20
Lista de Problemas Escassez de informações pré-natais;
Ecografias oriundas da UGO pouco descritivas; Oligoâmnio grave; RNP/PIG; IR grave; Instabilidade hemodinâmica; Orelhas com implantação baixa;

21
PATOLOGIA (filme da autópsia: Dr. Paulo R. Margotto)
")
22
Supra-renais Cortesia Dr. Paulo R. Margotto

23
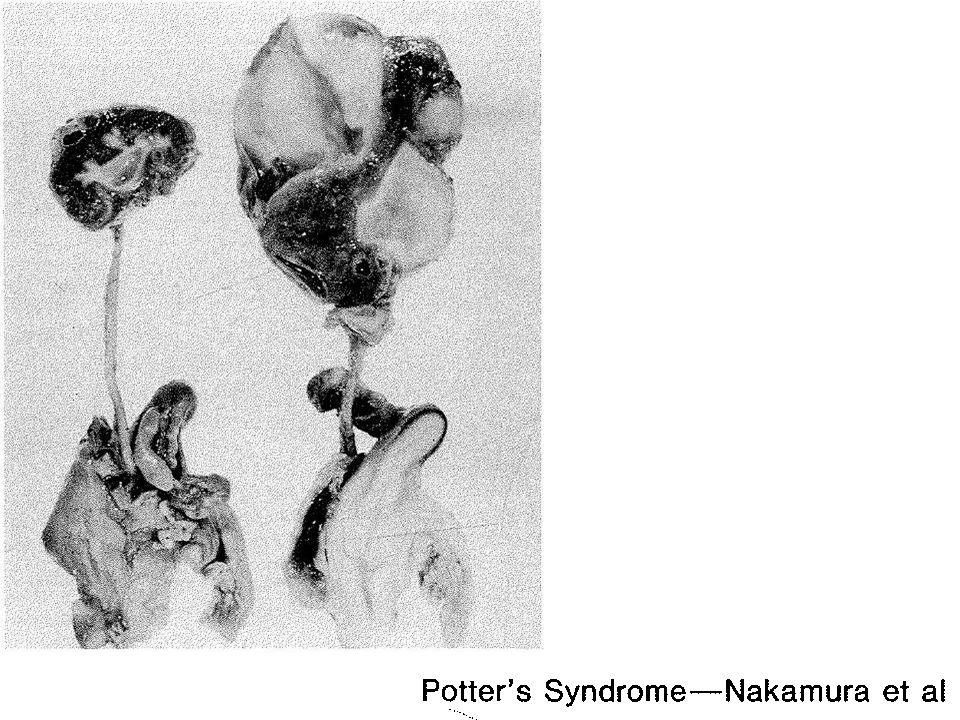
Ausência de rins e ureteres e bexiga rudimentar
Cortesia Dr. Paulo R. Margotto

24
Pulmões hipoplásicos Cortesia Dr. Paulo R. Margotto

25
Cortesia Dr. Paulo R. Margotto

26
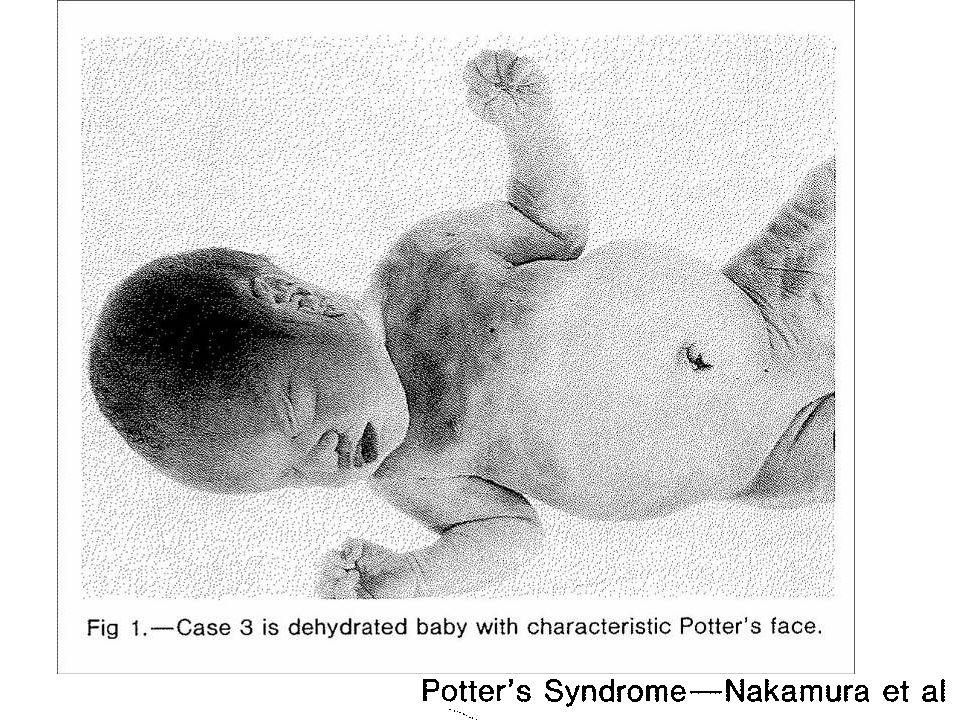
Síndrome de Potter

27
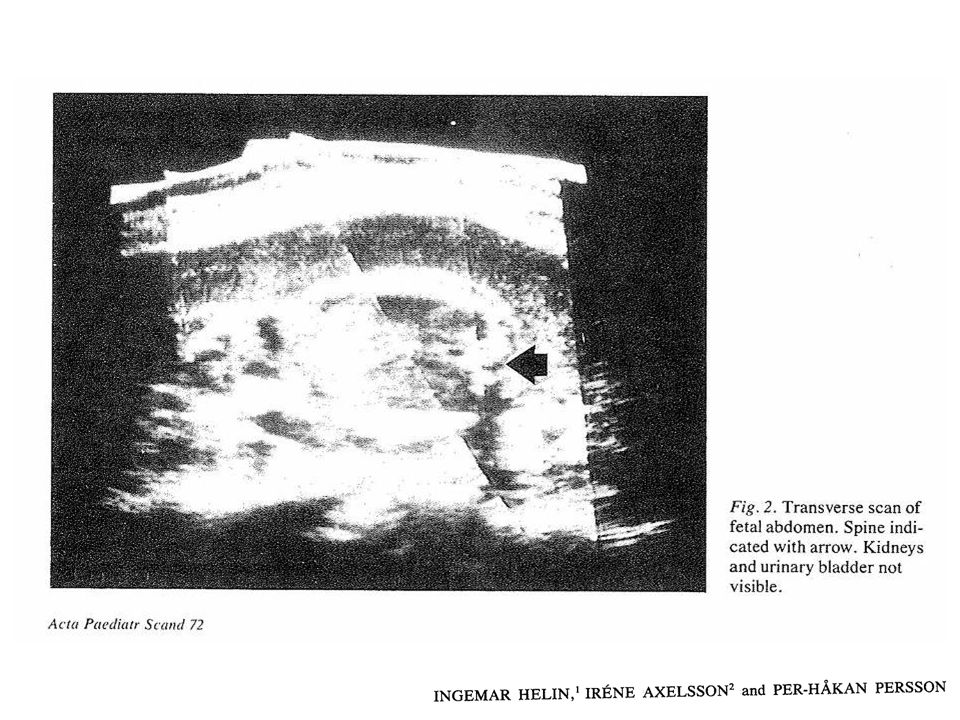
Correspondence to: P M Dunn, Department of Child Health, University of Bristol, Southmead Hospital, Southmead, Bristol BS10 5NB, UK; Dr Edith Potter (1901–93).
.")
28
1) Conceito Conjunto de achados fenotípicos fetais relacionados a oligoâmnio grave secundário à insuficiência renal.
 Conceito Conjunto de achados fenotípicos fetais relacionados a oligoâmnio grave secundário à insuficiência renal.")
29
2) Epidemiologia Incidência de 0 – 3/1000 nascimentos (Potter, 1946);
Proporção de 2 – 7/1 M/F; Baixo peso; PIG; Cariótipos geralmente normais; Apresentação pélvica é comum.

30
Pashayan, Dowd, and Nigro, 1977

31
Pashayan, Dowd, and Nigro, 1977

32
3) Etiologia ETIOLOGIA DESCONHECIDA;
Substâncias teratogênicas não encontradas; Causa genética: autossômica recessiva c/ sexo como fator limitante ? herança recessiva ligada ao sexo ? supressão 22q11 ?

33
Pashayan, Dowd, and Nigro, 1977

34
Agenesia renal Oligoâmnio Impactação Hipoplasia feto/parede Pulmonar
4) Fisiopatologia Agenesia renal Oligoâmnio Impactação Hipoplasia feto/parede Pulmonar
 Fisiopatologia. Agenesia renal Oligoâmnio Impactação Hipoplasia feto/parede Pulmonar")
35
Fitch and Lachance, 1972

36
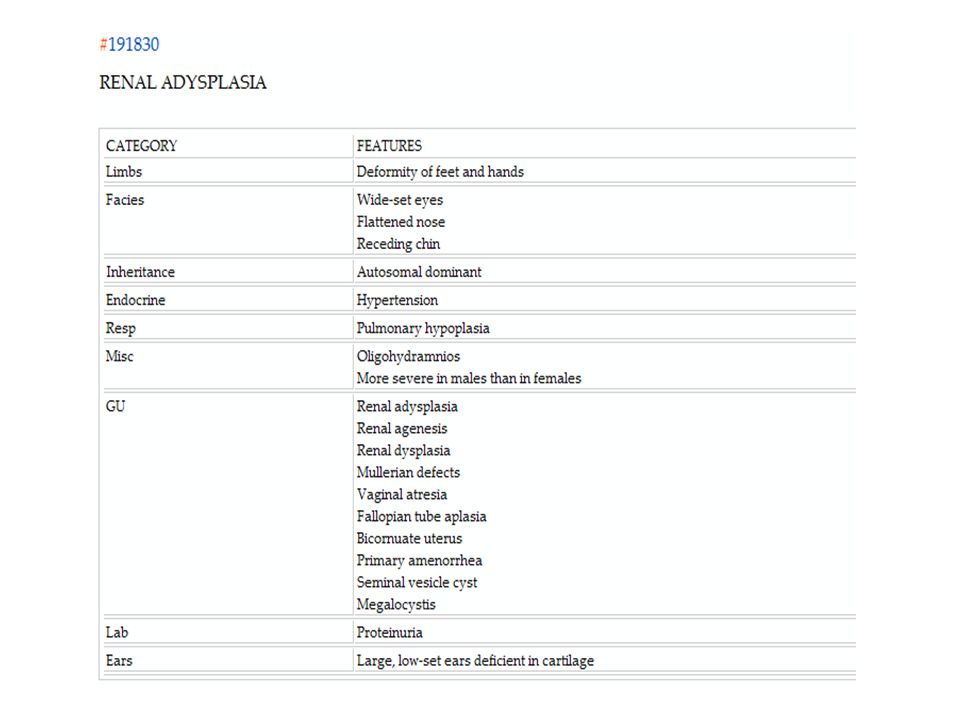
5) Quadro clínico Implantação baixa das orelhas; Hipertelorismo;
Pregas epicânticas Nariz achatado; Pescoço curto; Membros tortos; Insuficiência respiratória; Micrognatia; Pulmões rígidos durante reanimação.

37
Pashayan, Dowd, and Nigro, 1977

38
Fitch and Lachance, 1972

39
Pashayan, Dowd, and Nigro, 1977

40
Fitch and Lachance, 1972

42
Margarita et al, 2005

43
Trissomia do 7 A. Biri et al. / European Journal of Medical Genetics 48 (2005) 67–73
 67–73")
44
Trissomia do 7 A. Biri et al. / European Journal of Medical Genetics 48 (2005) 67–73
 67–73")
45
Fitch and Lachance, 1972

46
6) Patologia Agenesia renal bilateral; Hipoplasia pulmonar;
Ausência de líquido amniótico; Outras malformações associadas.

47
Margarita et al, 2005

49
Margarita et al, 2005

51
7) Curso da doença Natimortos: 60% dos casos; Morte em até 48 horas;
Evolução para IR em 2 semanas; Gestação geralmente termina antes do termo.

52
Quadro de prognóstico inevitavelmente fatal!
8) Tratamento Quadro de prognóstico inevitavelmente fatal!
 Tratamento. Quadro de prognóstico inevitavelmente fatal!")
53
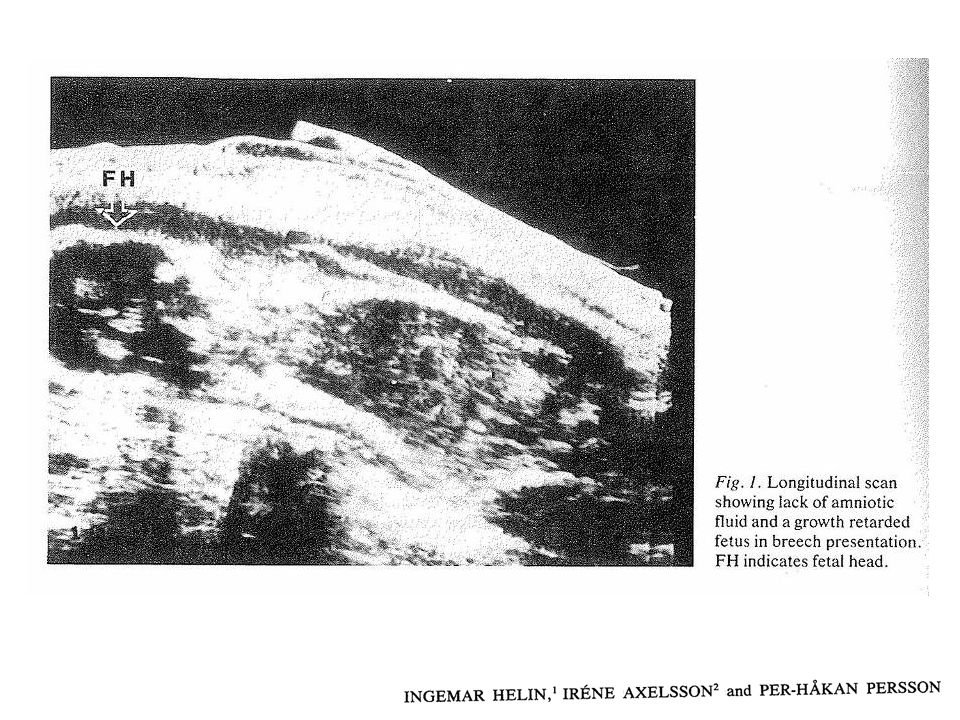
9) Prevenção Rastreamento ecográfico pré-natal;
Dificuldade ecográfica antes de 20 semanas de gestação; Desenvolvimento da anomalia no 1 trimestre; Oligoâmnio observado no final do 2 trimestre.

56
Margarita et al, 2005

57
10) Diagnóstico Diferencial
I) Sirenomelia SINDROME DE REGRESSÃO CAUDAL / SIRENOMELIA Autor(es): Zaw W, Stone D G/Das BB, Rajegowda BK, Bainbridge R, Giampietro P Cortesia Dr. Paulo R. Margotto
 Sirenomelia. SINDROME DE REGRESSÃO CAUDAL / SIRENOMELIA Autor(es): Zaw W, Stone D G/Das BB, Rajegowda BK, Bainbridge R, Giampietro P. Cortesia Dr. Paulo R. Margotto.")
58
Cisto em plexo coróide descritos em mais de 50% das autópsias, sendo geralmente menor que 1mm e não causam sintomas obstrutivos. Acredita-se que representam dobras neuroepiteliais que podem subsequentemente se encherem de líquor cefalorraquidiano e debris celulares. Estes cisto podem desaparecer por volta da semana incidência de ocorrência de cisto no plexo coróide de 3%, variando de 1-4mm, raramente ultrapassando 7mm (seguimento de 13 meses; sem alterações; pode ser um achado normal e não requer seguimento) Cisto de 2-8 cm podem ocasionar sintomas (hipertensão intracraniana) em crianças mais velhas e adultos, dependendo da sua localização (no foramen de Monro -fazer acompanhamento por 6 meses) Margotto,2011;Neurossonografia neonatal Fakhry et al, Lam e Villanueva, Riebel et al
 Cisto de 2-8 cm podem ocasionar sintomas (hipertensão intracraniana) em crianças mais velhas e adultos, dependendo da sua localização (no foramen de Monro -fazer acompanhamento por 6 meses) Margotto,2011;Neurossonografia neonatal. Fakhry et al, Lam e Villanueva, Riebel et al.")
59
Cisto no plexo coróide Ostlere et al tem relacionado grandes cistos bilaterais (>1mm) no plexo coróide com cromossopatia, principalmente trissomia do 18 (3%) e possivelmente, trissomia do 21. Fitzsimmons et al relataram que 71% dos fetos abortados com trissomia do 18 apresentavam cistos no plexo coróide No entanto, para submeter a paciente a diagnóstico invasivo, analisar a presença de restrição do crescimento intra-uterino e a presença de outras anormalidades estruturais. Margotto,2011;Neurossonografia neonatal
 no plexo coróide com cromossopatia, principalmente trissomia do 18 (3%) e possivelmente, trissomia do 21. Fitzsimmons et al relataram que 71% dos fetos abortados com trissomia do 18 apresentavam cistos no plexo coróide. No entanto, para submeter a paciente a diagnóstico invasivo, analisar a presença de restrição do crescimento intra-uterino e a presença de outras anormalidades estruturais. Margotto,2011;Neurossonografia neonatal.")
60
Cisto no plexo coróide Lam e Villanueva Margotto, Castro
enorme cisto no plexo coróide (setas) extendendo-se do teto do 3º ventrículo para o corno frontal do ventrículo esquerdo Margotto, Castro Margotto,2011;Neurossonografia neonatal
 extendendo-se do teto do 3º ventrículo para o corno frontal do ventrículo esquerdo. Margotto, Castro. Margotto,2011;Neurossonografia neonatal.")
61
Quando não iniciar/Quando interromper a reanimação
Requisitos para a não intervenção: Malformações facilmente reconhecidas; Prognóstico letal ou sobrevivência vegetativa; Diagnóstico laboratorial rapidamente disponível. Cariótipo, US, TC: Simples defeito requerendo correção heeóica ou medidas terapêuticas para sobrevida: agenesia renal bilateral. Margotto, PR, Goldsmith (1998)
")
62
Quando não iniciar/Quando interromper a reanimação
RN com severas anomalias congênitas; Distúrbios identificáveis ou defeitos específico-estruturais incompatíveis com a vida (Lista de Jones): Hidroanencefalia; Anencefalia; Trissomias do 13 e do 18; Agenesia renal bilateral; Holoprosencefalia; Triploidia; Sirenomelia; Síndrome dos membros curtos. Margotto, PR, Goldsmith (1998)
: Hidroanencefalia; Anencefalia; Trissomias do 13 e do 18; Agenesia renal bilateral; Holoprosencefalia; Triploidia; Sirenomelia; Síndrome dos membros curtos. Margotto, PR, Goldsmith (1998)")
63
Romina S. Heredia Garcia Silva Genética Médica (R2) 2011
ASPECTOS GENÉTICOS Romina S. Heredia Garcia Silva Genética Médica (R2) 2011
")
64
Síndrome: ocorre quando o agente responsável pela malformação (gene mutante ou teratógeno) causa múltiplas anormalidades em paralelo. Sequência: ocorre quando o agente responsável pela malformação afeta apenas um órgão em um ponto no tempo e é a perturbação desse órgão que provoca uma gama de defeitos secundários em outras estruturas. Nussbaum RL., McInnes R., Willard HF.Thomsom & Thompsom Genética Médica. Rio de Janeiro: Elsevier, 2008

65
Alterações renais no feto Perda crônica de LA
OLIGOÂMNIOS Compressão fetal Falta de desenvolvimento alveolar

67
Doenças renais perinatais letais: BRA BRD URA/RD SOU
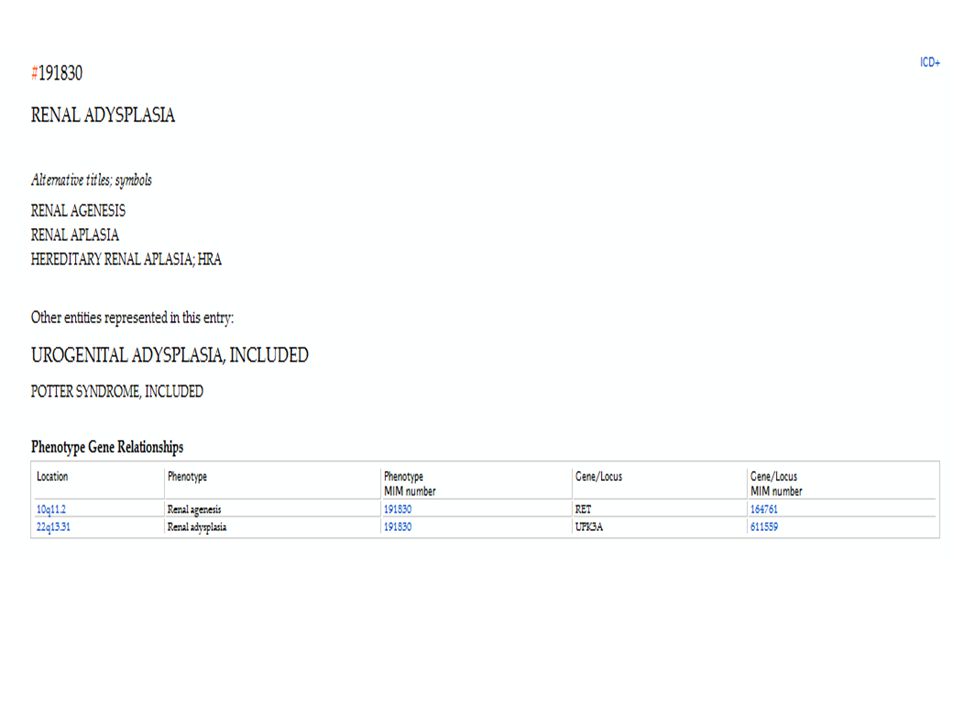
“Síndrome Potter” → Potter (1946) – alterações renais (incluindo hipoplasia renal e doença multicística) + “fácies de Potter”. Casos familiares – espectro fenotípico: adisplasia renal hereditária (Buchta et al., 1973)
 – alterações renais (incluindo hipoplasia renal e doença multicística) + fácies de Potter . Casos familiares – espectro fenotípico: adisplasia renal hereditária (Buchta et al., 1973)")
69
A alteração de renal não é patognomônica da síndrome Potter (Scott and Goodburn, 1995).
Outros autores relataram, ainda, outras alterações renais (Schmidt et al., Kiprov et al., 1982). McPherson et al. (1987): herança AD com penetrância entre 50-90%, com um risco empírico de 15-20% na descendência de um afetado ou heterozigoto obrigatório (adisplasia renal).
. McPherson et al. (1987): herança AD com penetrância entre 50-90%, com um risco empírico de 15-20% na descendência de um afetado ou heterozigoto obrigatório (adisplasia renal).")
70
Diagnóstico Ecografia gestacional Ecografia abdominal Necropsia
Ecografia renal dos pais e dos irmãos: 9% dos parentes em 1º grau apresentaram malformações renais assintomáticas. (Roodhooft, 1984) Outros... Citogenética [aberrações no cromossomo 5; monossomia 10q26; translocação (1;2)(q32;p25)]. Genética molecular: mutações nos genes UPK3A e RET
 Outros... Citogenética [aberrações no cromossomo 5; monossomia 10q26; translocação (1;2)(q32;p25)]. Genética molecular: mutações nos genes UPK3A e RET.")
71
Gene RET (protooncogene): receptor tirosina-kinase (molécula da superfície celular que traduz sinais para o crescimento e diferenciação celular). Mutãções no gene RET estão associadas a S. MEN2A, MEN2B, HSCR, MTC, agenesia renal (agenesia renal →perda de função, inativação da proteína – Skinner et al., 2008). Gene UPK3A (uroplakin 3A): proteína integral de membrana do urotélio. Mutações nesse gene resultariam em obstrução funcional ao fluxo urinário, levando a alterações no desenvolvimento dos rins (Jenkins et al., 2005).
: proteína integral de membrana do urotélio. Mutações nesse gene resultariam em obstrução funcional ao fluxo urinário, levando a alterações no desenvolvimento dos rins (Jenkins et al., 2005).")
72
Deltas C. ,Papagregoriou G. Cystic Diseases of the Kidney
Deltas C.,Papagregoriou G. Cystic Diseases of the Kidney. Arch Pathol Lab Med 134: , 2010.

75
CAKUT; genes mutated in the renal-coloboma (paired box 2; PAX2), renal cysts and diabetes (hepatocyte nuclear factor 1b; HNF1b) and branchio-oto-renal (eyes absent 1; EYA1) syndromes encode transcription factors and related proteins
, renal cysts and diabetes (hepatocyte nuclear factor 1b; HNF1b) and branchio-oto-renal (eyes absent 1; EYA1) syndromes encode transcription factors and related proteins")
76
“The identification of the exact nature of the lesion is therefore of paramount importance, not only for the prognosis of the propositus, but especially for the correct genetic counselling of the family.” (Moerman P, Fryns JP -2002).
.")
77
Bibliografia consultada
The pathogenesis of Potter's syndrome of renal agenesis (1972): Naomi Fitch, M.D., PH.D. and R. Claude Lachance, M.D., F.R.C.P. [C], Montreal; Double aneuploidy involving trisomy 7 with Potter sequence (2005): Aydan Biri, MeralYirmibes Karaog uz , Gönül DidemI˙nce , Mehmet Ali Ergün , Sevda Menevs, Banu Bingöl; Síndrome de Potter (2005): Hospital General “Dr. Juan Bruno Zayas” Síndrome de Potter Bilateral absence of the kidneys and ureters (1977): From the Department ofPediatrics, Tufts-New England Medical Center Hospitals, Boston Floating Hospitalfor Infants and Children, Boston, Massachusetts, USA Chromosome 22ql 1 deletion presenting as the Potter sequence (1997): Koenraad Devriendt, Philippe Moerman, Dominique Van Schoubroeck, Kamiel Vandenberghe, Jean-Pierre Fryns Prognosis of antenatally diagnosed oligohydramniosof renal origin (05/01/2007): Markus J. Kemper & Dirk E. Mueller-Wiefel PRENATAL DIAGNOSIS OF POTTER´S SYNDROME BY ULTRASOUND (1983): INGEMAR HELIN, IRÉNE AXELSSON and PER-HÂKAN PERSSON; Potter´s Syndrome Associated With Renal Agenesis or Dysplasia (1985); Dr Edith Potter (1901–1993) of Chicago: pioneer in perinatalpathology (2005). Margotto PR. Neurosonografia neonatal, ESCS, 2011 (no prelo)
: Naomi Fitch, M.D., PH.D. and R. Claude Lachance, M.D., F.R.C.P. [C], Montreal; Double aneuploidy involving trisomy 7 with Potter sequence (2005): Aydan Biri, MeralYirmibes Karaog uz , Gönül DidemI˙nce , Mehmet Ali Ergün , Sevda Menevs, Banu Bingöl; Síndrome de Potter (2005): Hospital General Dr. Juan Bruno Zayas Síndrome de Potter. Bilateral absence of the kidneys and ureters (1977): From the Department ofPediatrics, Tufts-New England Medical Center Hospitals, Boston Floating Hospitalfor Infants and Children, Boston, Massachusetts, USA. Chromosome 22ql 1 deletion presenting as the Potter sequence (1997): Koenraad Devriendt, Philippe Moerman, Dominique Van Schoubroeck, Kamiel Vandenberghe, Jean-Pierre Fryns. Prognosis of antenatally diagnosed oligohydramniosof renal origin (05/01/2007): Markus J. Kemper & Dirk E. Mueller-Wiefel. PRENATAL DIAGNOSIS OF POTTER´S SYNDROME BY ULTRASOUND (1983): INGEMAR HELIN, IRÉNE AXELSSON and PER-HÂKAN PERSSON; Potter´s Syndrome Associated With Renal Agenesis or Dysplasia (1985); Dr Edith Potter (1901–1993) of Chicago: pioneer in perinatalpathology (2005). Margotto PR. Neurosonografia neonatal, ESCS, 2011 (no prelo)")
78
Deltas C.,Papagregoriou G. Cystic Diseases of the Kidney. Arch Pathol Lab Med 134: , 2010. Nussbaum RL., McInnes R., Willard HF.Thomsom & Thompsom Genética Médica. Rio de Janeiro: Elsevier, 2008 Jones KL. Smith’s recognizable patterns of human malformation. Philadelphia: Elsevier,2006 Kerecuk L. et al. Autosomal dominant inheritance of non-syndromic renal hypoplasia and dysplasia: dramatic variation in clinical severity in a single kindred. Nephrol Dial Transplant (2007) 22: 259–263
 22: 259–263.")
79
O Quarteto Fantástico Dr. Paulo R. Margotto, Ddo Carlos, Ddo Éder e Ddo Thiago

Apresentações semelhantes






 Prognostic factors and survival in neonates with congenital.>")


>")
 MSS 22 anos, procura o serviço para fazer um cariótipo e receber aconselhamento genético. È G1, P1, A0, teve um filho pré termo.>")




:235-42>")



