Carregar apresentação
A apresentação está carregando. Por favor, espere

1
RNA-SEQ: CONCEITO E APLICAÇÕES
Disciplina BMP 5762 – Bioinformática Aplicada ao Estudo de Doenças Parasitárias RNA-SEQ: CONCEITO E APLICAÇÕES Ana da Rocha Kurata Katie Cristina Takeuti Riciluca

2
RNA-seq RNA-seq é uma abordagem recentemente desenvolvida, para analisar o perfil de transcriptoma, que utiliza tecnologias de deep-sequencing. O transcriptoma é o conjunto completo de transcritos (RNAs) em uma célula, e sua quantidade, para um estágio de desenvolvimento específico ou condição fisiológica. * deep-sequencing = indica que a cobertura do processo é muito maior que o comprimento da sequencia em estudo.
 em uma célula, e sua quantidade, para um estágio de desenvolvimento específico ou condição fisiológica. * deep-sequencing = indica que a cobertura do processo é muito maior que o comprimento da sequencia em estudo.")
3
O entendimento do transcriptoma é essencial para:
Interpretar os elementos funcionais do genoma Revelar os constituintes moleculares de células e tecidos nos diferentes estágios de desenvolvimento Compreender os elementos presentes no desenvolvimento de doenças O transcriptoma pretende catalogar todos os tipos de transcritos: mRNAs RNAs não codificadores pequenos RNAs.

4
Porquê estudar o transcriptoma?
Para determinar a estrutura transcripcional dos genes, em termos de seus sítios de início 5’ e final 3’; Padrões de splicing e outras modificações pós-traducionais; Quantificar os níveis de mudanças de expressão de cada transcrito durante o desenvolvimento e sob condições diferentes. Encontrar microRNAs que possuem função reguladora Metagenômica * Splicing = é um processo que remove os íntrons e junta os éxons depois da transcrição do RNA. O splicing só ocorre em células eucarióticas, já que o DNA das células eucarióticas não possui íntrons.

5
Criação da Biblioteca Pode-se utilizar: Todo o RNA da célula
Possui 90-95% de rRNA Apenas mRNA selecionado pela cauda de poli-A Perde-se microRNAs e mRNAs sem poli-A Retirando o rRNA Por hibridização com sequencias específicas ligadas a biotina que são retiradas com esferas ligadas a streptovidina Quebra por uma exonuclease que age sobre RNAs que possuem fosfato na extremidade 5' (apenas rRNAs possuem esse fosfato) A remoção de rRNAs aumenta a detecção e a montagem de transcritos raros. Mas se o objetivo do estudo é a quantificação, é necessário uma biblioteca não depletada.
 A remoção de rRNAs aumenta a detecção e a montagem de transcritos raros. Mas se o objetivo do estudo é a quantificação, é necessário uma biblioteca não depletada.")
6
Criação da Biblioteca Para a criação da biblioteca o RNA é transformado em cDNA por uma transcriptase reversa Para não se perder a direcionalidade do transcrito podem ser acrescentados adaptadores a uma extremidade do RNA isso é muito importante no estudo de espécies de genoma muito compactado onde o transcrito pode se sobrepor em fitas opostas O RNA pode ser fragmentado antes da formação de cDNA evitando a formação de estrutura secundária

8
Cada molécula de cDNA, com ou sem amplificação, é então sequenciada com um método de alto rendimento para obter sequências curtas de um final (sequenciamento single-end) ou de ambos os lados (sequenciamento pair-end). As leituras são tipicamente 30 – 400 bp, dependendo da tecnologia usada para sequenciamento do DNA. Para esse método tem se usado plataformas tipo: Illumina IG, SOLiD e 454.

9
Considerações Prioritárias na montagem
Para garantir uma alta qualidade na montagem do transcriptoma, cuidados particulares devem ser tomados nos experimentos de RNA-Seq. Na fase de análise de dados, as leituras curtas são pré-processadas para remover erros de sequenciamento e outros artefatos. As leituras são subsequentemente montadas nos RNAs originais e então sua abundância é avaliada.

10
[Martin, J. A.; Wang, Z. 2011]
![[Martin, J. A.; Wang, Z. 2011]](http://slideplayer.com.br/slide/4133701/12/images/10/%5BMartin%2C+J.+A.%3B+Wang%2C+Z.+2011%5D.jpg "[Martin, J. A.; Wang, Z. 2011]")
11
Para evitar erros na montagem de RNA, é necessário retirar o passo de amplificação por PCR
Na etapa de amplificação por PCR alguns fragmentos podem ser melhor amplificados que outros prejudicando os dados Já é possível fazer o sequenciamento sem amplificação usando as plataformas Helicos e Pacific Biosciences, O sequenciamento através de uma única molécula é possível, porém essas tecnologias ainda sofrem com a alta taxa de erro.

12
Estratégias de Montagem do Transcriptoma
Baseado em três categorias : Etratégia baseada em referência Estratégia de novo Estratégia combinada

13
Estratégia baseada em Referência
Quando existe um genoma de referência o transcriptoma pode ser construido a partir dele. Esse método inclui três passos: Alinhamento das leituras sobre o genoma de referência As leituras sobrepostas em cada locus são agrupadas para construir um gráfico de todas as isoformas possíveis. O gráfico é analisado para resolver isoformas individuais. Programas: Blat, TopHat, SpliceMap, MapSplice, GSNAP

14
[Martin, J. A.; Wang, Z. 2011]
![[Martin, J. A.; Wang, Z. 2011]](http://slideplayer.com.br/slide/4133701/12/images/14/%5BMartin%2C+J.+A.%3B+Wang%2C+Z.+2011%5D.jpg "[Martin, J. A.; Wang, Z. 2011]")
15
[Martin, J. A.; Wang, Z. 2011]
![[Martin, J. A.; Wang, Z. 2011]](http://slideplayer.com.br/slide/4133701/12/images/15/%5BMartin%2C+J.+A.%3B+Wang%2C+Z.+2011%5D.jpg "[Martin, J. A.; Wang, Z. 2011]")
16
Após as leituras serem alinhadas ao genoma, dois métodos são usados para a construção dos gráficos:
Cufflinks - cria um gráfico de sobreposição de todas as leituras que alinham com um único locus para montar isoformas encontrando o mínimo de transcritos que explicam os introns dentro da leitura. é mais conservativo na escolha de quais os transcritos são re-construidos Scripture - cria um gráfico que une cada base de um cromossomo e adiciona nas laterais (conexões) entre as bases se existe uma leitura que liga duas bases. pode produzir um grande conjunto de transcritos de um locus.
 entre as bases se existe uma leitura que liga duas bases. pode produzir um grande conjunto de transcritos de um locus.")
17
Vantagens Pode montar transcritos de baixa abundância;
Pode usar computação paralela Pode ser feita em máquinas com poucos gb de RAM; Descobrir novos transcritos que não estão em anotações já existentes; Descarta artefatos e contaminantes (que não alinham) Usado para transcriptomas simples: bactérias, archeaeal, eucarióticos simples com poucos introns pouco splicing alternativo
 Usado para transcriptomas simples: bactérias, archeaeal, eucarióticos simples. com poucos introns. pouco splicing alternativo.")
18
Desvantagens Não é possível sem um genoma de referência;
Depende da qualidade do genoma de referência ; Genomas podem não ser completos, ter regiões não agrupadas e parcialmente montadas. Genes que se encontram muito próximos ou sobrepostos podem ser interpretados com um único transcrito Não une leituras que esteja muito distantes no genoma ou em cromossomos diferentes

19
Estratégia de novo Não utiliza um genoma de referência;
Se utiliza da redundância das leituras para encontrar sobreposições entre as leituras Programas usam o gráfico De Brujin para reconstruir transcritos de uma ampla faixa de níveis de expressão e então processar a montagem de contigs e remover redundancias. Semelhante à montagem de genoma

20
[Martin, J. A.; Wang, Z. 2011]
![[Martin, J. A.; Wang, Z. 2011]](http://slideplayer.com.br/slide/4133701/12/images/20/%5BMartin%2C+J.+A.%3B+Wang%2C+Z.+2011%5D.jpg "[Martin, J. A.; Wang, Z. 2011]")
21
[Martin, J. A.; Wang, Z. 2011]
![[Martin, J. A.; Wang, Z. 2011]](http://slideplayer.com.br/slide/4133701/12/images/21/%5BMartin%2C+J.+A.%3B+Wang%2C+Z.+2011%5D.jpg "[Martin, J. A.; Wang, Z. 2011]")
22
Vantagens Não depende de um genoma de referência;
Pode providenciar um novo conjunto de dados de transcritos para genomas que não apresenta alta qualidade; Pode ser usado para encontrar transcritos exógenos ou que estão faltando no genoma; Não é influenciado por longos introns Encontra transcritos trans-spliced, resultantes de rearranjos cromossomais Pode ser utilizado para o transcriptoma de organismos complexos

23
Desvantagens A montagem de organismos eucariotos complexos pode consumir muita memória RAM Grande quantidade de dados Complexidade dos gráficos de Brujin nescessários para analizar os possíveis splicings Consome dias ou semanasde processamento Exige maior cobertura(30x) Suscetível a erros de leitura, pode não diferenciar um erro do sequenciamento de um splicing Trechos similares(como parálogos) ainda podem ser considerados um só transcrito
 Suscetível a erros de leitura, pode não diferenciar um erro do sequenciamento de um splicing. Trechos similares(como parálogos) ainda podem ser considerados um só transcrito.")
24
Estratégia Combinada A combinação dos dois métodos pode ser utilizada
O alinhamento tem a vantagem da sensibilidade O De Novo para encontrar transcritos novos e trans-spliced Realizando o alinhamento primeiro podemos descartar as sequências já conhecidas Fazendo a montagem De Novo com uma quantidade muito menor de dados Quando o genoma de referência tem baixa qualidade a montagem De Novo pode ser feita primeiro Os contigs e singlets são alinhados no genoma e as lacunas podem ser preenchidas com informações do genoma

25
[Martin, J. A.; Wang, Z. 2011]
![[Martin, J. A.; Wang, Z. 2011]](http://slideplayer.com.br/slide/4133701/12/images/25/%5BMartin%2C+J.+A.%3B+Wang%2C+Z.+2011%5D.jpg "[Martin, J. A.; Wang, Z. 2011]")
26
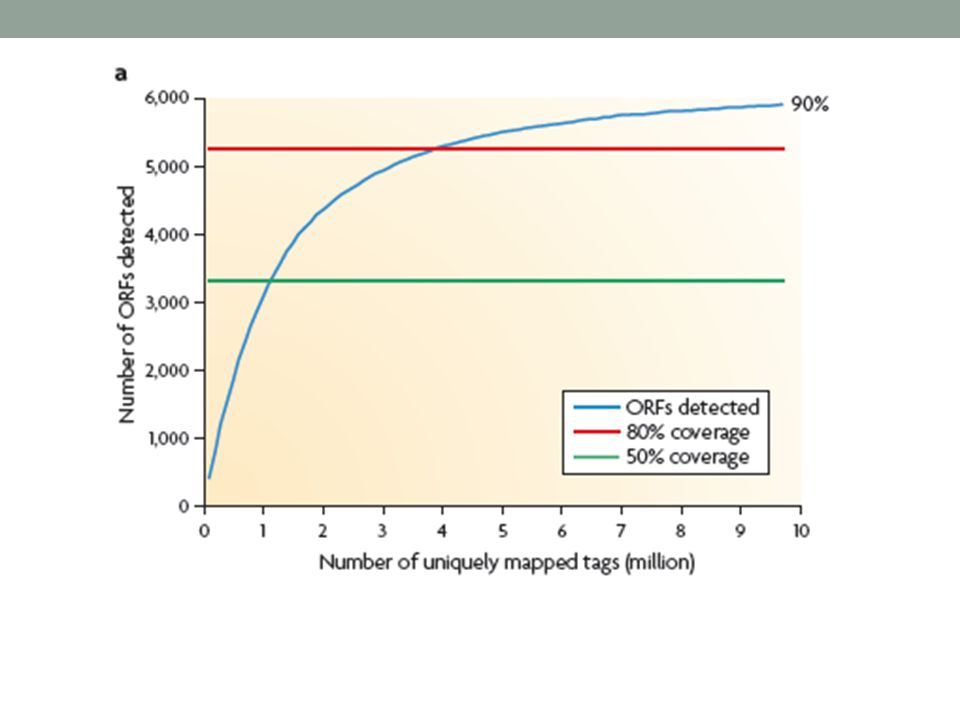
Cobertura x Custo Uma questão importante é a cobertura da sequência ou a porcentagem dos transcritos pesquisados, os quais implicam no custo. Grandes coberturas requerem mais sequenciamento. Em transcriptomas simples, como da levedura S. cerevisiae, que não tem evidência de splicing alternativo, 30 milhões de leituras de 35 nucleotídeos são suficientes para observar a transcrição de mais de 90% dos genes de células em crescimento sob uma condição unica

28
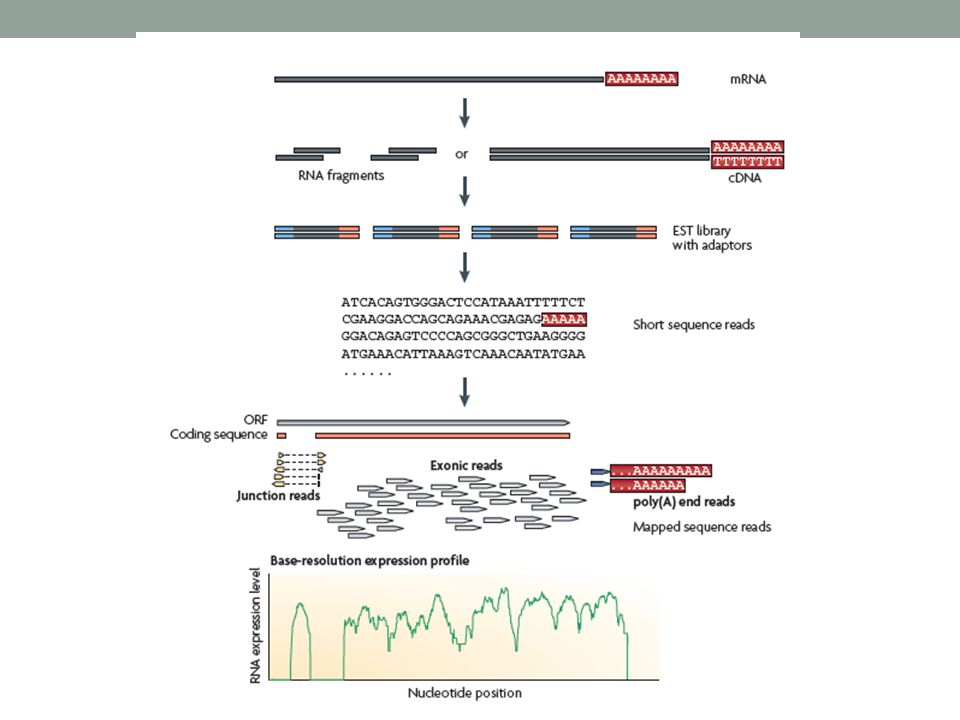
RNA-Seq revela a localização precisa dos limites da transcrição, com a resolução base a base.
Além disso, pequenas leituras de 30 pb de RNA-Seq nos mostra informação como 2 exons estão conectados, enquanto leituras longas ou leituras curtas por pair-ends poderiam revelar conectividade entre exons múltiplos. Os resultados de RNA-Seq também mostram alto nível de reprodutibilidade, para ambas as técnicas e replicatas biológicas.

29
Utilizações Descoberta de pequenos RNAs
Quantificação da expressão em diferentes momentos Fusão de genes em câncer Identificação de mutações Metagenômica

30
Obrigada!

Apresentações semelhantes





 dos genes e seu armazenamento>")













