Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Dissolução Desenvolvimento e Validação de Métodos
Margareth R. C. Marques, M.Sc., Ph.D Marco de 2004

2
Fontes de informação www.dissolutiontech.com www.dissolution.com
Chemical Abstracts

3
Importância do Teste de Dissolução
Pesquisa e Desenvolvimento direciona desenvolvimento e avaliação de novas formulações auxilia na avaliação da estabilidade de formulações permite correlação in-vivo/in-vitro auxilia na aprovação de um novo produto pelas autoridades regulatórias.

4
Importância do Teste de Dissolução
Controle de Qualidade / Produção faz parte das especificações do produto detecção de desvios de produção assegurar consistência e reprodutibilidade dentro de um mesmo lote e lote a lote avaliação da qualidade da formulação em função do tempo e condições de armazenagem avaliação de modificações no processo auxilia na decisão sobre estudos de bioequivalência

5
Quando fazer dissolução
Comprimidos: mastigável, desintegração oral, revestidos, sublingual, que não desintegram, etc Cápsulas: duras, moles, com conteúdo líquido Suspensões, granulados para suspensões Supositórios Pomadas, géis, cremes Adesivos transdérmicos Implantes Lipossomas, nanosferas, etc Gomas de mascar

6
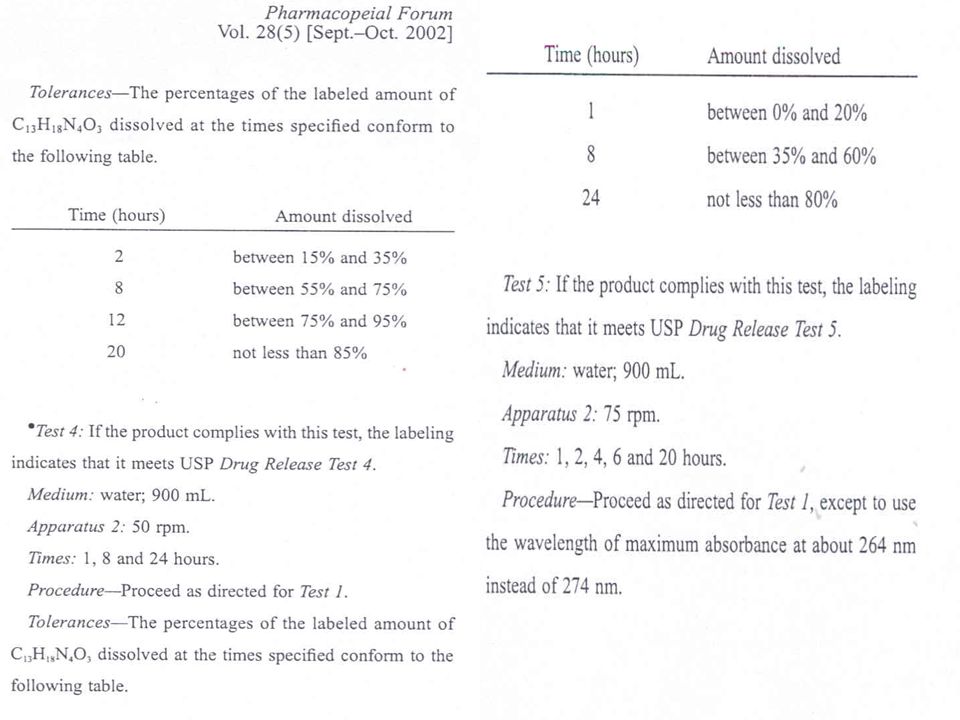
USP - Processo de Revisão
Pharmacopeial Forum Publicação bimensal Contem propostas de novas monografias, capitulos gerais ou modificacoes

28
Monografias USP Formas Farmaceuticas de liberacao imediata
Grande chance teste USP funcionar Validacao Formas farmaceuticas de liberacao modificada Pequena chance teste USP funcionar Caso a caso – mecanismo de liberacao Desenvolvimento e validacao

29
Proposta de novo capítulo geral
Gray, V. A., Brown, C. K., Dressman, J. B., Leeson, L. J. - A new general information chapter on dissolution. Pharm. Forum 27(6): , 2001. <1092> The Dissolution Procedure: Development and Validation. Pharm. Forum 30(1): , 2004
: , <1092> The Dissolution Procedure: Development and Validation. Pharm. Forum 30(1): ,")
30
Desenvolvimento de métodos
Seleção de condições meio de dissolução aparelho Método discriminativo preciso transferível robusto

31
Método discriminativo
Capacidade de detectar mudanças no produto Ideal - mudanças que afetam comportamento in vivo - IVIVC Testar formulações fabricadas com parâmetros diferentes fármaco formulação processo de fabricação

32
Método discriminativo
Fármaco tamanho de partícula - área de superfície forma cristalina - polimorfos solvatação rota de síntese Formulação diferentes quantidades de excipientes granulação úmida x seca

33
Método discriminativo
Processo de fabricação tempo de mistura força de compressão ordem de adição método de secagem método de revestimento equipamento

34
Desenvolvimento de métodos
Condições atípicas podem ser utilizadas, MAS: devem ter justificativa resultados comparativos mostrando vantagens

35
Teste de Dissolução Reflete mudanças formulação processo de fabricação
características físico-químicas do fármaco tamanho de partícula área de superfície polimorfismo estado de hidratação

36
Teste de Dissolução Mudanças afetam
solubilidade desempenho in vivo do produto Mudança no comportamento de dissolução pode ou não afetar a performance in vivo do produto

37
Teste de Dissolução Estudo de estabilidade refletir os efeitos de
temperatura tempo umidade degradação fotosensibilidade

38
Teste de Dissolução Equipamento
deve estar de acordo com as especificações e deve atender os requisitos de verificação do sistema descritos nos capítulos gerais <711> Dissolution e <724> Drug release da USP .

39
Teste de Dissolução Equipamento USP 1 - cesto Equipamento USP 2 - pás
cápsulas e comprimidos agitação 50 a 100 rpm Equipamento USP 2 - pás Cápsulas, comprimidos e suspensoes agitação 50 ou 75 rpm

40
Velocidade de agitação
Entre 25 a 150 rpm Inaceitável fora dessa faixa Abaixo de 25 rpm - hidrodinâmica não reprodutível Acima de 150 rpm - turbulência Entre 25 e 50 rpm - aceitável para suspensões

41
Velocidade de agitação
Formação de cone no fundo da cuba a 50 rpm. Cone pode ser reduzido aumentado velocidade para 75 rpm, ou até 100 rpm. Produto dissolve muito rápido ou muito lento - diminuição ou aumento da velocidade de agitação.

42
Equipamento Formas Farmacêuticas de Liberação Modificada
mesmos requisitos que para liberação imediata escolha baseada na formulação e no mecanismo de liberação in vitro Equipamento USP 1 ou 2 Aumento na velocidade de agitação

43
Equipamento Formas Farmacêuticas de Liberação Modificada
Equipamento USP 3 (cilindros recíprocos) - adequada para formulações do tipo grânulo ou pérola. Equipamento USP 4 (célula de fluxo) - princípios ativos com baixa solubilidade em solventes aquosos.
 - adequada para formulações do tipo grânulo ou pérola. Equipamento USP 4 (célula de fluxo) - princípios ativos com baixa solubilidade em solventes aquosos.")
44
Equipamento USP 3 (cilindros recíprocos)
Forma farmacêutica

45
Equipamento USP 4 (célula de fluxo)
")
46
Equipamento USP 4 Permite velocidades de fluxo superiores a 50 mL/min (3 L/h) Uso com implantes Sistema fechado ou aberto

47
Sistema aberto Flow Cell Fresh Medium Piston Pump

48
Sistema fechado com espectrofotômetro em linha
Splitter Spectrophotomter with Cell-Changer Flow Cell Magnetic Stirrer Piston Pump

49
Equipamento Formas Farmacêuticas de Liberação Modificada
Equipamento USP 5 (pá sobre disco) e Aparelho USP 6 (cilindro) - transdérmicos. Equipamento USP 7 (disco recíproco) - formas sólidas orais que não desintegram e transdérmicos.
 e Aparelho USP 6 (cilindro) - transdérmicos. Equipamento USP 7 (disco recíproco) - formas sólidas orais que não desintegram e transdérmicos.")
50
Equipamento USP 5 (pá sobre disco)
Forma farmacêutica coberta com tela

51
Equipamento USP 6 (cilindro rotatório)
")
52
Equipamento USP 7 (disco recíproco)
Suporte Forma farmacêutica

53
Equipamento USP 7 (disco recíproco)
")
54
Comprimidos que não desintegram - bombas osmóticas

55
Comprimidos que não desintegram - bombas osmóticas

56
Adesivos transdérmicos

57
Adesivos transdérmicos

58
Equipamento Cesto Abertura da malha uniforme
40 mesh outras aberturas podem ser utilizadas (10 ou 20 mesh) uniforme dimensões de acordo com <711> Dissolution entupimento - usar pás
 uniforme. dimensões de acordo com <711> Dissolution. entupimento - usar pás.")
59
Equipamento Cuba com pico - presença de cone
Mini pás - em combinação com cuba de 200 mL - produtos com baixa dosagem Cubas de 2 L ou 4 L Outros equipamentos Frascos rotatorios (microparticulados injetaveis ou implantes) Outras modificações
 Outras modificações.")
60
Âncoras Utilizadas em conjunto com Equipamento USP 2
cápsulas e comprimidos que flutuam flutuação geralmente observada no início do teste centralização da forma farmacêutica embaixo da pá comprimidos revestidos ou cápsulas de gelatina mole que aderem à parede da cuba.

61
Âncora - J. Pharm.

62
Âncoras

63
Meio de dissolução Dados químicos e físicos do fármaco e da forma farmacêutica Fármaco solubilidade estabilidade em solução em função do pH influência de tensoativos

64
Forma farmacêutica dureza friabilidade
presença de agentes solubilizantes excipientes que afetam dissolução revestimentos velocidade de desintegração mecanismo de liberação

65
Meio de dissolução Escolha em função da solubilidade do fármaco
doses do produto “sink condition” deve ser atendida

66
“Sink condition” Mínimo três vezes o volume de meio de dissolução necessário para obter solução saturada do fármaco.

67
Meio de dissolução Faixa fisiológica de pH: 1,2 a 6,8 (1,2 a 7,5 para formas modificadas) fármaco instável nesse meio - justificar escolha pH deve ser medido antes e depois do teste para verificar se houve mudanças.

68
Meio de dissolução Misturas com solventes orgânicos - devem ser evitadas Água - não ideal qualidade variável pH variável ausência de capacidade tamponante Tensão superficial variável e dependente dos excipientes presentes

69
Meio de dissolução Ácido clorídrico (0,1 N a 0,001 N)
Tampão acetato (pH 4,1 a 5,5 - 0,05 M) Tampão fosfato (pH 5,8 a 8,0 - 0,05 M) Água purificada Solução polisorbato 20, 40, 60, 80 Solução lauril sulfato de sodio Solução óxido de laurildimetilamina Solução cetrimida
 Tampão fosfato (pH 5,8 a 8,0 - 0,05 M) Água purificada. Solução polisorbato 20, 40, 60, 80. Solução lauril sulfato de sodio. Solução óxido de laurildimetilamina. Solução cetrimida.")
70
Meio de dissolução Solução de sais biliares e/ou lecitina
Combinação de tensoativos e ácidos ou tampões Fluido gástrico simulado com ou sem enzimas Fluido intestinal simulado com ou sem enzimas Preparação de acordo com a seção da USP “Reagents, Indicators, and Solutions”.

71
Meio de dissolução Avaliar a estabilidade do fármaco no meio escolhido. Fármacos com baixa solubilidade em solventes aquosos Uso de tensoativos. Justificar concentração e tipo

72
Meio de dissolução Meio biorelevante
Meio que tem alguma relevância nas condições de dissolução in vivo do fármaco em análise. Considerar sitio de absorção fator limitante: dissolução ou permeabilidade

73
Meio de dissolução Fármaco dissolve rapidamente no estômago e é rapidamente absorvido condições gástricas - pH ácido Dissolução ocorre no intestino pH básico - fluido intestinal simulado pH 6,8

74
Meio de dissolução Meios que simulam estado de jejum e após as refeições estabelecer correlação in vivo/in vitro avaliar efeito dos alimentos avaliar mecanismo de liberação não tem utilização em CQ

75
Meio de dissolução Uso de enzimas
de acordo com capítulo geral USP <711> Dissolution formação de película de gelatina cápsulas de gelatina ou comprimidos revestidos com gelatina

76
Meio de dissolução Volume 500 mL a 1000 mL, 900 mL mais comum
volume 2 ou 4 L “sink condition”

77
Meio de dissolução Desaeração
presença de bolhas interferem no resultado bolhas agem como barreira à dissolução bolhas facilitam a aderência de partículas tanto na cuba como no mecanismo de agitação bolhas na superfície da forma farmacêutica aumentam a flutuação causando aumento da dissolução

78
Meio de dissolução Desaeração
realizada de acordo com capítulo geral USP <711> Dissolution outros métodos, desde que validados meios contendo tensoativos não são desaerados.

79
Retirada da Amostra

80
Zona de amostragem (900 mL)
Amostrar na metade da distância entre a superfície do meio e o topo do elemento agitador. Não menos que 1 cm da parede lateral da cuba.

81
Filtração

82
Tempo e especificações
Forma Farmacêutica de Liberação Imediata típico 30 a 60 minutos ponto único Quantidade liberada: porcentagem do teor declarado no rótulo (Q) típico 70% a 80% acima de 80% não são utilizados. Considerar teor e uniformidade da dose. Perfil de dissolução
 típico 70% a 80% acima de 80% não são utilizados. Considerar teor e uniformidade da dose. Perfil de dissolução.")
83
Perfil de dissolução Porcentagem do fármaco dissolvido versus tempo
pontos selecionados para caracterizar a parte ascendente e o platô do perfil fármacos altamente soluvéis e permeáveis: um ponto único, tipicamente 80% ou 85% em 15 min típico: 70% - 80% em min

84
Perfil de dissolução Pontos a 15, 20, 30, 45 e 60 min
5 a 10 min para fármacos que dissolvem rapidamente acima de 60 min para fármacos com dissolução lenta

85
Perfil de dissolução Ponto infinito
após o último ponto do perfil de dissolução, velocidade de 150 rpm por 30 a 60 min informação complementar à uniformidade de distribuição da dose características de desempenho da formulação

86
Tempo e especificações
Formas Farmacêuticas de Liberação Modificada mínimo 3 pontos Ponto inicial - 1 ou 2 horas Ponto intermediário - define perfil Ponto final - completa liberação do fármaco

87
Tempo e especificações
Produtos contendo mais que um princípio ativo Dissolução é determinada para cada um dos componentes Possivelmente com critérios de aceitação diferentes

88
Observações visuais Características da formulação
Variações no processo de produção aderência das partículas formação de cone partículas flutuando na superfície do meio presença de bolhas oclusão da malha do cesto Forma farmaceutica se movendo

89
Variabilidade nos resultados
Problemas se: desvio padrão relativo >20% em 10 min desvio padrão relativo >10% nos outros tempos Causas mais comuns formulação artefatos associados com o teste adesão do produto ao copo ou mecanismo de agitação

90
Variabilidade nos resultados
Resultados aberrantes se o conteúdo da forma farmacêutica não está disperso de maneira uniforme. Mudança de aparelho ou velocidade desaeração tipo de âncora composição do meio

91
Variabilidade nos resultados
Formulação dose não uniforme inconsistências no processo reação ocorrendo durante a dissolução interação com excipientes revestimentos envelhecimento da cápsula endurecimento/amolecimento

92
Validação de Métodos de Dissolução

93
Validação Amostragem Seleção de filtros e âncoras Desaeração
Especificidade - interferência dos excipientes Faixa de linearidade Exatidão/Recuperação Precisão Robustez Estabilidade das soluções

94
Amostragem Manual Ponto de amostragem - <711>
seringas plásticas ou de vidro cânula de aço inox filtro suporte para filtro Ponto de amostragem - <711> Amostragem através do cesto ou da haste tem que ser validado

95
Amostragem Manual x Automática contaminação cruzada
adsorção do fármaco efeito da presença de sonda de amostragem ciclos de limpeza e enxague

96
Amostragem manual x automática
Resultados de dissolução não são altamente variáveis (RSD < 20% a 10 min, RSD < 10% nos outros tempos) Dois testes de dissolução (n = 6, cada). Um com amostragem manual e o outro com automática Perfil de dissolucao Correção do volume nos cálculos Comparação dos resultados através dos critérios de Precisão Intermediária
 Dois testes de dissolução (n = 6, cada). Um com amostragem manual e o outro com automática. Perfil de dissolucao. Correção do volume nos cálculos. Comparação dos resultados através dos critérios de Precisão Intermediária.")
97
Amostragem manual x automática
Resultados de dissolução são altamente variáveis (RSD > 20% a 10 min, RSD > 10% nos outros tempos) Amostragem simultânea da mesma cuba a cada ponto do perfil de dissolução Perfil de dissolucao Correção do volume nos cálculos Comparação dos resultados através dos critérios de Precisão Intermediária
 Amostragem simultânea da mesma cuba a cada ponto do perfil de dissolução. Perfil de dissolucao. Correção do volume nos cálculos. Comparação dos resultados através dos critérios de Precisão Intermediária.")
98
Seleção do filtro Interromper a dissolução
Remover excipientes insolúveis Deve ser pré umedecido com o meio de dissolução 0,45 µm a 70 µm Adsorção do fármaco deve ser avaliada

99
Filtros Solução padrão a 100% filtrada comparada com solução padrão não filtrada Solução da amostra (cuba de dissolução ou béquer) filtrada comparada com solução centrifugada Resultados devem estar entre 98% e 102% da concentração original das soluções não filtradas
 filtrada comparada com solução centrifugada. Resultados devem estar entre 98% e 102% da concentração original das soluções não filtradas.")
100
Uso de âncoras Descrição do tipo utilizado
Justificativa para utilização Comparação com diferentes tipos, teste com cada um Habilidade em manter a forma farmacêutica no fundo do copo Grande influência no perfil de dissolução Caso a caso Transferência de método

101
Desaeração Verificação da necessidade de desaerar ou não o meio de dissolução. Comparação dos resultados com meio desaerado e não desaerado.

102
Especificidade - Influência dos excipientes
Placebo: todos os excipientes incluindo revestimentos 3 amostras de cada mistura placebo, equivalentes às doses maior e menor adição dos revestimentos, pigmentos e âncora, cápsula de gelatina, etc Transferir para a cuba contendo meio de dissolução a 37°C Agitar por 30 a 60 min a 150 rpm utilizando o aparelho descrito no método

103
Especificidade - Influência dos excipientes
Comparação com solução padrão equivalente a 100% da dose Calcular a porcentagem de interferência para cada dose (média de 3) Interferência não deve ser maior que 2%.
 Interferência não deve ser maior que 2%.")
104
Especificidade - Influência dos excipientes
Produtos com liberação modificada Uso de placebo ao invés de mistura de excipientes Placebo mesmo mecanismo de liberação que o produto Interferência determinada a cada tempo de amostragem no perfil de dissolução

105
Especificidade - Influência dos excipientes
Se a interferência dos excipientes excede 2% comprimentos de onda alternativos método analítico alternativo uso de espectrofotometria com segunda derivada fator de correção

106
Especificidade Doseamento por HPLC
presença de picos eluindo no mesmo tempo que o fármaco presença de picos estranhos - injete solução padrão e compare tempos de retenção Tempos de retenção muito próximos, contamine a solução do placebo com o fármaco

107
Especificidade Doseamento por HPLC
Correr o cromatograma com o branco (meio de dissolução), solução padrão e solução da amostra por um certo período de tempo para verificar presença de picos que eluem mais tarde. Cromatogramas de meio de dissolução, solução do placebo filtrada, solução padrão, amostra de dissolução filtrada.
, solução padrão e solução da amostra por um certo período de tempo para verificar presença de picos que eluem mais tarde. Cromatogramas de meio de dissolução, solução do placebo filtrada, solução padrão, amostra de dissolução filtrada.")
108
Especificidade Ausência de picos interferentes no cromatograma do placebo Ausência de absorbância do placebo no comprimento de onda do teste.

109
Faixa de Linearidade Máximo 5% (v/v) de solvente orgânico para aumentar a solubilidade do padrão nas soluções. Outras quantidades devem ser validadas. Cinco soluções padrão variando de ±20% da dose menor até ±20% da dose mais alta que será liberada durante o teste. Leituras no mínimo em duplicata Maior concentração não deve exceder os limites de linearidade do equipamento.
 de solvente orgânico para aumentar a solubilidade do padrão nas soluções. Outras quantidades devem ser validadas. Cinco soluções padrão variando de ±20% da dose menor até ±20% da dose mais alta que será liberada durante o teste. Leituras no mínimo em duplicata. Maior concentração não deve exceder os limites de linearidade do equipamento.")
110
Faixa de linearidade Cálculos utilizando programa de regressão linear de quadrados mínimos. r2 0.98 Intercessão com o eixo y deve ser significativamente diferente de zero com 95% de limite de confiança

111
Exatidão / Recuperação
Em geral ±20% da concentração mais baixa até ±20% da concentração mais alta que será liberada durante o teste. Mínimo três níveis de concentração em triplicata. Incluir cápsula de gelatina, revestimentos, pigmentos, âncora, etc. Quantidade de placebo a cada nível deve ser a mesma que o total de excipientes da dose em análise.

112
Exatidão / Recuperação
Equipamentos 1 e 2 - testar a mistura de excipientes e fármaco nas condições especificadas no método. Fármacos com baixa solubilidade - solução estoque - fármaco dissolvido em solvente orgânico (máx 5% vol final), volume completado com meio de dissolução. Solução estoque diluída ou transferida para a cuba. Recuperação 95% a 105% das quantidades pesadas.
, volume completado com meio de dissolução. Solução estoque diluída ou transferida para a cuba. Recuperação 95% a 105% das quantidades pesadas.")
113
Exatidão / Recuperação
Formas farmaceuticas de liberacao retardada – Revestimento gastroresistente Estagio acido Limite “maximo 10% liberado” deve ser validado

114
Precisão Reprodutibilidade réplicas das solução padrão
cálculo do desvio padrão relativo

115
Precisão Precisão intermediária
efeito de eventos aleatórios na precisão do método determinada dentro da faixa de linearidade do método no mínimo 2 analistas em diferentes dias uso de lote bem caracterizado, faixa estreita de uniformidade da dose

116
Precisão intermediária
Perfil de dissolução da mesma amostra em duplicata Determinado por no mínimo dois analistas Cada analista preparando suas próprias solução padrão e meio Diferente equipamentos Diferente dias

117
Precisão intermediária
Diferença no valor médio obtido por um analista em relação ao outro analista não é maior que 10% (absoluto) nos tempos com dissolução menor que 85% Diferença máxima de 5% para os tempos com dissolução acima de 85% Critério de aceitação pode ser específico do produto
 nos tempos com dissolução menor que 85% Diferença máxima de 5% para os tempos com dissolução acima de 85% Critério de aceitação pode ser específico do produto.")
118
Robustez Avaliada durante o desenvolvimento do teste
Concentração do meio Composicao do meio pH do meio Método quantitativo

119
Estabilidade das soluções
Solução padrão Analisada durante um período específico de tempo Comparação com solução recém preparada a cada intervalo de tempo Resultados entre 98 e 102% do valor teórico

120
Estabilidade das soluções
Solução da amostra Armazenada a temperatura ambiente Analisada durante um período específico de tempo Comparação com o resultado inicial Resultados entre 98% e 102% do valor inicial

121
Estabilidade das soluções
Caso necessário, o método deve indicar, no texto, prazo de validade das soluções. Verificada utilizando outros métodos analíticos (HPLC, UV/Vis).
.")
122
Referências Bibliográficas
Gray, V. A., Brown, C. K., Dressman, J. B., Leeson, L. - A new general information chapter on dissolution. Pharmacopeial Forum 27(6): , 2001. Nickerson, B. - Challenges and trends in dissolution testing. Am. Pharm. Rev., April 2001. Hokanson, G. C. - Setting specifications for solid oral dosage forms: leveraging development experience. Am. Pharm. Rev., 4(2), 2001. Rohrs, B. R. - Dissolution method development for poorly soluble compounds. Dissol Techn., August 2001.
: , Nickerson, B. - Challenges and trends in dissolution testing. Am. Pharm. Rev., April Hokanson, G. C. - Setting specifications for solid oral dosage forms: leveraging development experience. Am. Pharm. Rev., 4(2), Rohrs, B. R. - Dissolution method development for poorly soluble compounds. Dissol Techn., August")
123
Referências Bibliográficas
Noory, C., Tran, N., Ouderkirk, L., Shah, V. - Steps for development of a dissolution test for sparingly water-soluble drug products. Dissol. Techn. February 2000. Qureshi, S. A., Shabnam, J. - Cause of high variability in drug dissolution testing and its impact on setting tolerances. Eur. J. Pharm. Sci., 12: , 2001. Scott, P. Investigating out of specifications dissolution results: a systematic approach. Am. Pharm. Rev. Lozano, R., Agorrody, M., Beraud, B., Dauphin, J.F. - Gelatin cross-linking in capsules and its effect on dissolution. Am. Pharm. Rev.

124
Referências Bibliográficas
Singh, S., Rao, K. V. R., Venugopal, K. Manikandan, R. - Alteration in dissolution characteristics of gelatin-containing formulations. A review of the problem, test methods, and solutions. Pharm. Technol., April 2002,

125
Contato Margareth R. C. Marques, M.Sc., Ph.D. e-mail mrm@usp.org
tel fax

Apresentações semelhantes



















>")
: Karen Cristine Bergamo Nabarro Professor.>")












