Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Marina Macagnan Martha Arnold Paula Golin Rafael Soares Roberta Rohde Saulo Mabilde

2
Era uma vez...

3
No seu consultório... Uma criança de 1 ano de idade é trazida devido a retardo no crescimento A mãe reclama que a pele de seu filho parece “amarelada” Ela e seu marido são primos oriundos da Sardenha no Mediterrâneo Ela refere que vários familiares em seu país de origem têm “sangue fino” e que as vezes precisam de “injeções especiais”

4
Você notou que a criança:
estava abaixo do desenvolvimento esperado estava ictérica apresentava deformidade no crânio seu maxilar era bem pronunciado tinha esplenomegalia

5
Os exames mostraram que:
A criança tem anemia hemolítica severa com níveis reduzidos de hemoglobina no sangue Eritrócitos são deformados, pequenos, e mostram anormalidades estruturais. ELETROfORESE DA HEMOGLOBINA O sangue contém níveis aumentados de hemoglobina fetal- Hb F

6
O Rx de crânio mostrou o padrão “cabelo-em-escovinha” associado com aumento do espaço medular

7
?

8
-talassemia

9
Etiologia Histórico: - Von Jakhs - Thomas Cooley - Origem do Nome
Definição de talassemia Definição de β-talassemia Características Iniciais

10
Epidemiologia Distribuição geográfica : - origem
- mutações talassêmicas - movimentos migratórios Mutações com efeitos semelhantes: Países mediterrâneos África tropical Sudeste da Ásia Índia Sul da China

11
Epidemiologia Grandes Correntes Migratórias: - tráfico negreiro
- migração italiana - migração de caribenhos e africanos - indianos, cipriotas e paquistaneses - asiáticos Diagnóstico Intra-uterino Dados da O.M.S.: α-talassemia β-talassemia

12
Epidemiologia Β-talassemia pelo mundo: - Itália : 2 a 15 %
- Grécia : 8 % - Chipre : 18 % - Península Ibérica : 0,1 a 2 % - Sul da Europa : mais de 4 milhões de heterozigotos. - Am. Latina e Caribe : 1 a 2 % de heterozigotos.

13
Apresentação Clínica Três tipos diferentes de apresentação fenotípica
TALASSEMIA MAIOR - anemia de Cooley - reduzida síntese de HbA - dados clínicos: 1°. ano de vida (características peculiares) anemia intensa (hemoglobina<6,5 g/dl) esplenomegalia volumosa redução da massa muscular alterações craniofaciais atraso no crescimento
 anemia intensa (hemoglobina<6,5 g/dl) esplenomegalia volumosa. redução da massa muscular. alterações craniofaciais. atraso no crescimento.")
14
Apresentação Clínica obs.: transfusões crônicas de sangue
crianças tratadas inadequadamente ou tardiamente não tratado

15
Apresentação Clínica TALASSEMIA INTERMEDIÁRIA
β-talassemia heterozigótica mutações e combinações complexas manifestações clínicas: - anemia leve á moderada(hemoglobina entre 7 e 11 g/dl) - esplenomegalia - redução da massa muscular - alterações faciais - úlceras crônicas na perna - sintomas compressivos
 - esplenomegalia. - redução da massa muscular. - alterações faciais. - úlceras crônicas na perna. - sintomas compressivos.")
16
Apresentação Clínica TALASSEMIA MENOR
β-talassemia heterozigótica Uma única mutação β-talassêmica Dados clínicos: - anemia moderada a mínima - microcitose profunda - normalmente assintomáticos Obs.: diminuição dos níveis de hemoglobina diagnóstico difícil de ser estabelecido

17
Fisiologia

18
Síntese da Hemoglobina
Duas cadeias de globina ( e “não ”) Com exceção das primeiras semanas de embriogênese, uma cadeia é sempre . Feto: cadeia não chamada Depois do nascimento: cadeia β se pareia com a globina. Hemoglobina completa: 2 cadeias α e+ 2 cadeias não .
 Com exceção das primeiras semanas de embriogênese, uma cadeia é sempre . Feto: cadeia não chamada Depois do nascimento: cadeia β se pareia com a globina. Hemoglobina completa: 2 cadeias α e+ 2 cadeias não .")
19
Hemoglobina A

20
Síntese da Hemoglobina
Genes que codificam a cadeia da α globina estão no cromossomo 16. Genes que codificam a produção de cadeias não estão no cromossomo 11.

21
A Hemoglobina e seus genes

22
Ontogenia da Síntese da Hemoglobina
No cromossomo 11 os genes das globinas são ativados em seqüência durante o desenvolvimento, da extremidade 5´para 3´. ε embriogênese desenvolvimento fetal perto do nascimento

23
Ontogenia da Síntese da Hemoglobina

24
Hemoglobinas no Adulto
2 cadeias α + 2 cadeias β Hb A (97%) 2 cadeias α + 2 cadeias Hb A2 (2%) 2 cadeias α + 2 cadeias Hb F (1%)
 2 cadeias α + 2 cadeias Hb A2 (2%) 2 cadeias α + 2 cadeias Hb F (1%)")
25
Fisiopatologia

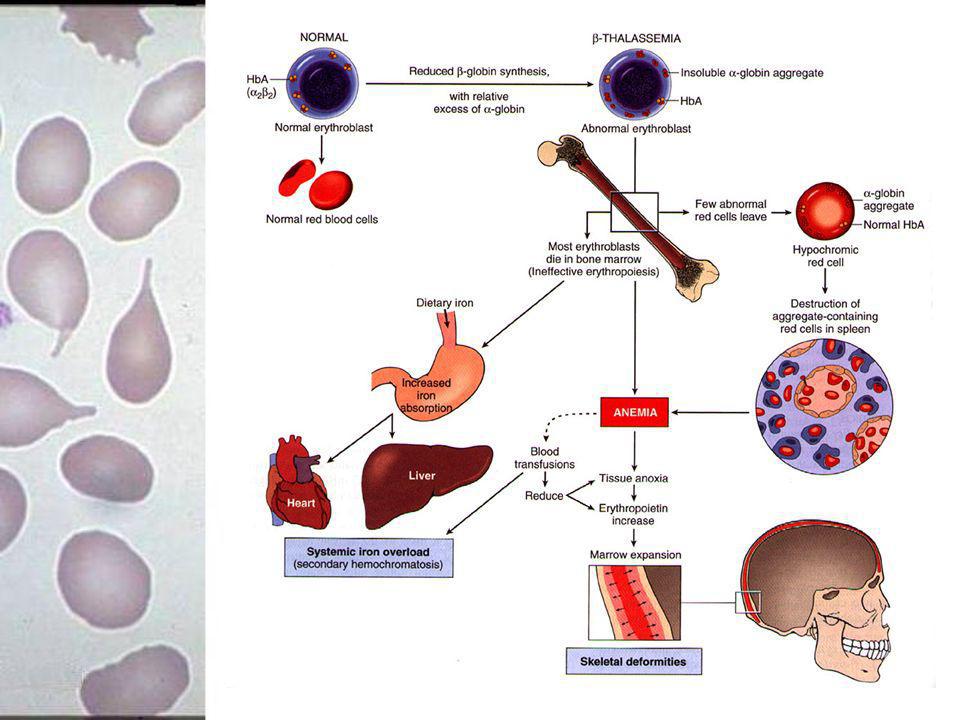
26
Fisiopatologia Produção de β globina: anemia microcítica hipocrômica
globina : importância só no período pós- natal beta talassemia evidente meses após nascimento

27
Fisiopatologia Cadeias em excesso insolúveis precipitação lesão membrana perda potássio síntese de DNA prejudicada destruição de precursores eritrocitários na MO (eritropoiese ineficaz) 70 a 85 % dos normoblastos podem ser destruídos Gene intacto níveis elevados
 70 a 85 % dos normoblastos podem ser destruídos. Gene intacto níveis elevados.")
28
Fisiopatologia Nível de Hb F aumentado devido à sobrevida seletiva e por produção aumentada da população de hemácias adultas que contém Hb F.

29
Alterações Morfológicas
Expansão da MO adelgaçamento do osso cortical neoformação óssea na face externa do osso (maxilar, ossos frontais da face e crânio) Hepatoesplenomegalia devido à hiperplasia do SRE e à hematopoiese extramedular. Hemossiderose, Hemocromatose secundária (transfusões, absorção intestinal de ferro)
 Hepatoesplenomegalia devido à hiperplasia do SRE e à hematopoiese extramedular. Hemossiderose, Hemocromatose secundária (transfusões, absorção intestinal de ferro)")
30
Alterações Morfológicas

31
Mecanismos da Anemia Eritropoiese pode aumentar + de 10 vezes, 95% pode ser ineficaz. Efeitos deletérios do excesso relativo de cadeias de globina. O excesso interfere na maturação eritropoiética normal: morte intramedular de precursores das células vermelhas por parada na fase G1; apoptose intramedular acelerada dos eritroblastos tardios

32
Mecanismos da Anemia Acúmulo de cadeias na membrana das células vermelhas: anormalidades bioquímicas danosas ao citoesqueleto Presença de ferro na membrana pode agravar efeitos deletérios.

33
Conseqüências clínicas da Anemia
Expansão da medula vermelha : 30X > que o normal volume plasmático (shunting através da medula expandida) e a progressiva esplenomegalia exacerbam a anemia. eritropoetina formação tecido extramedular massas extra-ósseas no abdome tórax e pelve
 e a progressiva esplenomegalia exacerbam a anemia. eritropoetina formação tecido extramedular massas extra-ósseas no abdome tórax e pelve.")
34
Conseqüências clínicas da Anemia
Expansão MO deformidades do crânio e face, osteopenia e defeitos na mineralização óssea podem agravar uma síndrome periarticular dolorosa microfraturas e osteomalácia.

36
Genética Molecular da -talassemia

37
Produto cadeia de β-globina humana
SEQ.CAP PROMOTOR EXON1 EXON2 SEQ 5´UTR INTRON1 INTRON2 EXON3 CODON TERM. 3´UTR Gene da β-Globina Humana (HBB) Locus 11p15.5 Produto cadeia de β-globina humana Presente no cluster do gene HBB, juntamente com os genes da γ-globina, ε-globina e δ-globina.
 Locus 11p15.5. Produto cadeia de β-globina humana. Presente no cluster do gene HBB, juntamente com os genes da γ-globina, ε-globina e δ-globina.")
38
CACCC proximal (motif)
PROMOTOR 5 Regiões de regulação tecido-específicas: CACCC proximal (motif) CACCC distal CAAT TATA GATA I
 CACCC distal. CAAT. TATA. GATA I.")
39
Recebe um cap nucleotídeos necessário para o RNAm
SEQ.CAP PROMOTOR ACATTTG Início da transcrição Recebe um cap nucleotídeos necessário para o RNAm ligar-se ao ribossomo para subseqüente tradução.

40
Seqüência transcrita e posteriormente não traduzida
SEQ.CAP PROMOTOR SEQ 5´UTR Seqüência transcrita e posteriormente não traduzida Importante para a estabilidade do RNA

41
SEQ.CAP PROMOTOR EXON1 SEQ 5´UTR 90 pares de bases aminoácidos 1-30

42
importante no processamento do RNAm nuclear e saída do núcleo
SEQ.CAP PROMOTOR EXON1 SEQ 5´UTR INTRON1 130 pares de bases importante no processamento do RNAm nuclear e saída do núcleo

43
SEQ.CAP PROMOTOR EXON1 EXON2 SEQ 5´UTR INTRON1 130 pares de bases aminoácidos

44
SEQ.CAP PROMOTOR EXON1 EXON2 SEQ 5´UTR INTRON1 INTRON2 850 pares de base

45
SEQ.CAP PROMOTOR EXON1 EXON2 SEQ 5´UTR INTRON1 INTRON2 EXON3 126 pares de bases aminoácidos

46
SEQ.CAP PROMOTOR EXON1 EXON2 SEQ 5´UTR INTRON1 INTRON2 EXON3 CÓDON TERM. Códon de terminação

47
Transcrito e não traduzido Sítio de ligação da Poli(A) do RNAm
SEQ.CAP PROMOTOR EXON1 EXON2 SEQ 5´UTR INTRON1 INTRON2 EXON3 CODON TERM. 3´UTR Transcrito e não traduzido Sítio de ligação da Poli(A) do RNAm
 do RNAm.")
48
RNAm nuclear RNAm maduro
Adição de um CAP de guanosina-metilada na extremidade 5´ Adição da cauda POLI(A), na extremidade 3´ CAP EXON1 EXON2 INTRON1 INTRON2 EXON3 POLI(A) RNAm maduro Processamento dos introns (splice) CAP EXON1 EXON2 EXON3 POLI(A)
, na extremidade 3´ CAP. EXON1. EXON2. INTRON1. INTRON2. EXON3. POLI(A) RNAm maduro. Processamento dos introns (splice) CAP. EXON1. EXON2. EXON3. POLI(A)")
49
Sumário dos passos envolvendo a produção da β-globina e da hemoglobina

50
Base Molecular da ß-Talassemia
São classificadas em , , , , , e dependendo da cadeia ou cadeias cuja síntese é defeituosa As -talassemias podem ser divididas de acordo com a ausência completa ou apenas deficiência na síntese de cadeia de -globina humana em: - 0-talassemia - +-talassemia

51
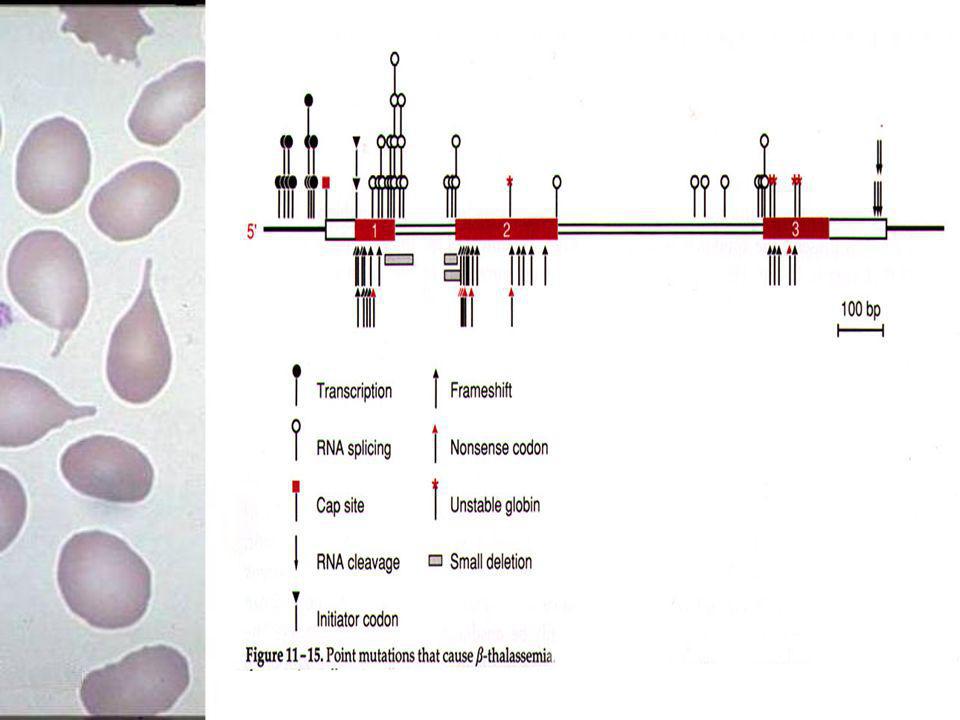
β-talassemias simples
Bases moleculares das β-talassemias simples A maior parte das mutações causam diminuição na produção de mRNA ou formação de mRNA não funcionais Um pequeno grupo de mutações, nonsense ou frameshift, ocorrem na região codificante do gene (exons) Grandes deleções no cluster do gene HBB podem resultar em talassemias complexas
 Grandes deleções no cluster do gene HBB podem resultar em talassemias complexas.")
52
Mutações que podem causar:
Classificação das mutações beta-talassêmicas Mutações que podem causar: Defeito na síntese de mRNA: splice junctions; introns; exons; Formação de mRNA não funcionais; Alteração na adição da cauda poli-A ou no caping; Mudança em áreas regulatórias.

53
Mutações nas splice junctions
Intron Exon Podem causar alteração nas regiões doadora 5’ou aceptora 3’ dos introns ou em seqüências consensuais que rodeiam as junções; Ocorre a formação de locais de splice crípticos, que nada mais são do que áreas alternativas de splice, que competem com áreas normais; Fenótipo é de β+-talassemia.

54
Mutações em Introns Intron Exon Qualquer mutação que torne uma região críptica de splice mais parecida ou até mesmo idêntica à região de splice normal Ocorre então competição entre a região críptica ativada e a região de splice normal Fenótipo de β+-talassemia

55
Mutações em Exons afetando o Splicing
Intron Exon Causam alteração na região codificante do gene HBB Podem resultar em mudança tanto na seqüência de aminoácidos da cadeia de β-globina quanto na quantidade de formação de RNAm específicos Fenótipo de variante estrutural. Exemplo HB E

56
Splicing

57
Mutações que causam formação de RNAm não funcional
Exon Exemplo são mutações que formam um código de parada prematuro Podem ser causadas por substituição de um único aminoácido ou por frameshift

58
Mutações que causam alteração na cauda Poly(A) ou no capping do RNAm
Impedem as modificações pós-transcricionais do mRNA, ou pelo capping não correto na extremidade 5’ ou por não poliadenização da extremidade 3’ Afetam a estabilidade do mRNA formado Fenótipo geralmente de β+-talassemia

59
Mutações que Afetam a Área do Promotor
Geralmente deleções ou mutagênese pontual O fenótipo depende dos boxes (seqüências consensuais) onde ocorreu a mutação No CACCC principal proximal fenótipo mais severo, enquanto no CACCC distal o fenótipo pode ser silencioso
 onde ocorreu a mutação. No CACCC principal proximal fenótipo mais severo, enquanto no CACCC distal o fenótipo pode ser silencioso.")
61
Mutação Fenótipo Origem Étnica
Promoter Mutantes -101 (C to T) B(+) Turquia -88 (C to A) B(+) Mediterrnea -87 (C to G) B(+) Negro Americana Mutantes que causam códon de parada Códon 1 (-1 bp) B(0) Chinesa Códon 6 (-1 bp) B(0) Mediterrânea Códon 114 (-2, +1 bp) B(+) Francesa Mutações na Splice Junction IVS-1, position 1 (G to A) B(0) Mediterrânea IVS-1, position 2 (T to G) B(0) Indiana, Chinesa Novo Local de Splice IVS (G to A) B(+) Mediterrânea Defeito no Cleavage do RNA AATAAA to AACAAA B(+) Negro Americana
 B(+) Turquia. -88 (C to A) B(+) Mediterrnea. -87 (C to G) B(+) Negro Americana. Mutantes que causam códon de parada. Códon 1 (-1 bp) B(0) Chinesa. Códon 6 (-1 bp) B(0) Mediterrânea. Códon 114 (-2, +1 bp) B(+) Francesa. Mutações na Splice Junction. IVS-1, position 1 (G to A) B(0) Mediterrânea. IVS-1, position 2 (T to G) B(0) Indiana, Chinesa. Novo Local de Splice. IVS (G to A) B(+) Mediterrânea. Defeito no Cleavage do RNA. AATAAA to AACAAA B(+) Negro Americana.")
62
Fatores Genéticos que Afetam a Severidade Clínica:
Natureza das mutações no gene HBB; Co-herança de -talassemia; Quantidade de hemoglobina fetal produzida.

63
LCR – Locus Control Region
Super-enhancer Estabelecem a abertura da cromatina, inibindo a repressão normal da transcrição Localizado aproximadamente 20 Kb anteriormente ao cluster Contém cinco locais que são hipersensíveis à DNase, apenas nas células eritróides precursoras

64
LCR – Locus Control Region
A presença do LCR inteiro é fundamental para a transcrição dos genes presentes no cluster do HBB Pacientes com deleções nos LCR falham em expressar todos os genes do cluster do HBB Essencial para terapia gênica

65
Padrão de Herança Autossômico recessivo;
Recentemente foi relatado em algumas famílias padrão de herança autossômico dominante.

66
Fenótipos Talassemia Menor Talassemia Maior Normal

67
Padrão de herança recessivo
Se ambos os pais não forem portadores então todos os seus filhos serão normais.

68
Padrão de herança recessivo
Se apenas um dos pais é portador e o outro normal, os filhos terão 50% de chance de ter Talassemia menor, porém nenhum deles terá Talassemia maior.

69
Padrão de herança recessivo
Se ambos os pais forem portadores, seus filhos terão 25% de chance de herdarem talassemia maior; 50% talassemia minor e 25% de chance de nascerem normais.

70
Padrão de herança autossômica dominante
Resultam de uma família de mutações, geralmente envolvendo o exon 3 do gene HBB Ocorre produção de cadeias de globina hiper-instáveis, com tamanhos variados, que se precipitam junto com as cadeias em em excesso de -globina nas células precursoras hemáticas

71
Diagnóstico

72
Diagnóstico Clínico Diagnóstico Clínico-laboratorial Eletroforese Cromatografia Análise Molecular Diagnóstico Diferencial

73
Diagnóstico Clínico História médica individual
- idade de início clínico História médica familiar Origem étnica Exame físico - crescimento e desenvolvimento - palidez e icterícia - tamanho hepático e esplênico - estrutura óssea

74
Diagnóstico Clínico-laboratorial
Hemograma completo -VCM -HCM / CHCM -RBC -RDW Esfregaço de sangue periférico

75
Diagnóstico Clínico-laboratorial
-VCM- Elemento <72 fL diagnóstico presuntivo

76
Diagnóstico Clínico-laboratorial
-HCM/CHCM- Talassemia menor : g/dL Talassemia maior : <7 g/dL

77
Diagnóstico Clínico-laboratorial
-RBC- Na anemia microcítica Talassemia RBC X Ferropenia RBC

78
Diagnóstico Clínico-laboratorial
-RDW- Talassemia : RDW X Ferropenia : RDW

79
Diagnóstico Clínico-laboratorial
Esfregaço de sangue periférico - microcitose - hipocromia - anisocitose - poiquilocitose - eritroblastos - inclusões de cadeias α

80
Esfregaço

81
Esfregaço

82
Eletroforese Objetivo: qualificar e quantificar a Hb - Hb A - Hb F

83
Eletroforese Normal + o Hb A (%) 97.5 73 0 Hb F (%) 0.8 24 97.5
 Hb F (%)")
84
Cellulose Acetate Electrophoresis
Pouca distinção entre Hb A e Hb F

85
Citrate Agar Electrophoresis
Teste confirmatório e pós-natal

86
Thin Layer Isoelectric Focusing
Gradiente de pH e boa resolução

87
Cromatografia Objetivo: maior precisão
HPLC - High Performance Liquid Cromatography Screening Custo elevado

88
HPLC-cromatografia

89
HPLC-cromatografia

90
Análise molecular Objetivo: identificar mutações específicas
Diferencia alelos Embasar aconselhamento genético Pós-diagnóstico clínico-laboratorial/pré-natal

91
Análise molecular PCR Seqüenciamento (eletroforese)
")
92
Diagnóstico Diferencial
Talassemia Anemia Ferropriva HEMOGRAMA X

93
Diagnóstico Diferencial
Mentzer index: VCM/RBC <13 talassemia >13 ferropenia Eletroforese perde valor

94
Tratamento

95
Tratamento da -talassemia Primeiro passo é distinguir entre:
Talassemia Maior Talassemia Menor Talassemia Intermediária

96
Tratamento da -talassemia Maior Transfusão Sangüínea
Terapêutica Quelante Transplante de Medula Óssea Esplenectomia Apoio Psicológico

97
Tratamento da -talassemia Maior Transfusão Sangüínea
Objetivo: HB > 10gdL Quando: precocemente Efeitos Favoráveis: crescimento, atividade física, hiperplasia medula óssea, deformidades ósseas, esplenomegalia Precauções: vacinação contra a Hepatite B, controle do sangue transfundido

98
Tratamento da -talassemia Maior Terapêutica Quelante
Objetivo: evitar hemossiderose Quando: após 1 ano do início das transfusões / após transfusões Como: infusão subcutânea lenta Efeitos Favoráveis: evitar lesões orgânicas graves como insuficiência de glândulas endócrinas, fígado e miocárdio provocadas pelo acúmulo de ferro Efeitos Colaterais: raros Uso da vitamina C associada

99
Transplante de Medula Óssea
Tratamento da -talassemia Maior Transplante de Medula Óssea Objetivo: cura Quando: antes que apareçam as complicações Como: irmão(ã) HLA idêntico Sucesso: 80% Riscos
 HLA idêntico. Sucesso: 80% Riscos.")
100
Tratamento da -talassemia Maior Esplenectomia
Objetivo: medida auxiliar Quando: benefícios maiores que a manutenção do baço Precauções: vacina anti-pneumococo Antibioticoterapia profilática Febre

101
Acompanhamento do Tratamento
Tratamento da -talassemia Maior Acompanhamento do Tratamento exame físico mensal ALT bimensal ferritina sérica a cada 3 meses crescimento e desenvolvimento audiometria e ofltalmo anualmente avaliação cardíaca completa, US fígado, tireóide, pâncreas, adrenal, paratireóide e hipófise

102
Tratamento da -talassemia Maior Apoio Psicológico
Esclarecer a natureza da doença, sua evolução, tratamento e complicações Reforçar a terapia quelante Adolescência Prognóstico Transfusões regulares Ferritina sérica abaixo de 2500 ng/mL

103
Tratamento Sintomático
Tratamento da -talassemia Intermediária Tratamento Sintomático Esplenectomia Suplementação ácido fólico Radioterapia de massas extramedulares Transfusões – quelantes Úlceras maleolares

104
Geralmente assintomático
Tratamento da -talassemia Menor Geralmente assintomático Confusão Gravidez

105
Tratamento Experimental da -talassemia
Três estratégias: Transferência de Genes Correção da seqüência DNA/RNA mutante Diminuição da expressão do gene da -globina

106
Diagnóstico Pré-natal

107
Objetivos Sistematizar o tratamento Melhorar prognóstico

108
Exames-padrão PCR e seqüenciamento de DNA: amniocentese
Eletroforese ou HPLC: cordocentese ou coleta de sangue fetal

109
Aconselhamento Genético
Método População de Risco Exames Aconselhamento Genético

110
Desvantagens Impossibilidade de coleta
Aborto espontâneo em 1-2% dos casos

111
Balanço Apesar de haver riscos, há grandes vantagens em fazer-se uso do diagnóstico pré-natal

112
Rastreamento

113
Definição Método pelo qual se busca, numa população de risco, indivíduos portadores de uma doença

114
Eficácia -talassemia: informação e métodos rápidos e acessíveis para realizar o rastreamento

115
Diagnóstico Neo-natal
Teste do pezinho

116
Método Métodos hematológicos e análise de DNA

117
Resultados Aparentemente o status de portador não causa ansiedade
Redução do nascimento de crianças beta-talassêmicas no Mediterrâneo

118
Aconselhamento Genético

119
Definição O aconselhamento genético é o processo de providência individual e familiar de informações sobre a natureza, hereditariedade e implicações de doenças genéticas para ajudá-los a tomar decisões informadas de caráter médico e pessoal

120
Objetivo Esclarecer: - Evolução da doença - Riscos para futuros filhos
Obs.: Não é objetivo do aconselhamento genético a erradicação da doença

121
Influências Inúmeras Modulam o caráter do serviço
Podem, mas não devem, mudar o desfecho

122
Enfoques Depende de: População Família Pessoa Exemplos

123
Princípio A decisão final sempre é do paciente e não deve ser influenciada pelo orientador do serviço

124
Benefício Prevenção de novos casos de -talassemia, pela prevenção de gravidezes de risco

125
Obrigado

Apresentações semelhantes