Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Aula 2 - Cinética Química
QFL 0464

2
O que iremos ver… O que é a cinética química?
Como poderemos avaliar uma reacção do ponto vista cinética Qual o impacto da temperatura da cinética da reacção Teoria da colisão - relação com a velocidade de reacção.

3
A Cinética química é que estuda a velocidade com que ocorrem de as reacções químicas.
Cinética Movimento ou Mudança Energia cinética é a energia associada ao movimento de um objecto Em Química - A palavra cinética refere-se à variação de concentração de um reagente ou de um produto com o tempo (e.g. M/s)
")
4
Existem muitas razões para estudar a velocidade de uma reacção.
Curiosidade intrínseca sobre a razão pela qual as reacções têm velocidades tão diversas. visão, fotossíntese têm uma escala de tempo na ordem de s Outras como o passagem da grafite ao diamante ocorrem na escala dos milhões de anos… Isto antes dos Russos produzirem diamantes artificiais de melhor qualidade que os diamantes naturais…

5
Reagentes produtos 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=− ∆ 𝐴 ∆𝑡 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒= ∆ 𝐵 ∆𝑡 AB
Em geral é mais conveniente exprimir a velocidade de uma reacção em termos da variação da concentração com o tempo 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=− ∆ 𝐴 ∆𝑡 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒= ∆ 𝐵 ∆𝑡 Onde ∆ 𝐴 𝑒 ∆ 𝐵 são variações de concentração durante o intervalo de tempo ∆𝑡

6
Não! Velocidade em cada um destes instantes é diferente
Será que podemos dizer que a velocidade da reacção é constante? Mas quando comparamos reacções nós queremos comparar dentro do mesmo intervalo de tempo para podermos afirmar que esta reacção é mais rápida que esta ou que a outra… Por outro lado, este gráfico diz-nos que a velocidade da reacção varia à medida que a concentração das espécies químicas envolvidas na reacção (reagentes e .produtos) varia…Então significa que deve existir uma correlação entre velocidade e concentração. Esta correlação pode ser avaliada pela lei das velocidades. Não! Velocidade em cada um destes instantes é diferente
 varia…Então significa que deve existir uma correlação entre velocidade e concentração. Esta correlação pode ser avaliada pela lei das velocidades. Não! Velocidade em cada um destes instantes é diferente.")
7
𝑎𝐴+𝑏𝐵→𝑐𝐶+𝑑𝐷 Lei da velocidade 𝑽𝒆𝒍𝒐𝒄𝒊𝒅𝒂𝒅𝒆=𝜥 𝑨 𝒙 𝑩 𝒚
Constante de velocidade 𝑽𝒆𝒍𝒐𝒄𝒊𝒅𝒂𝒅𝒆=𝜥 𝑨 𝒙 𝑩 𝒚 ORDEM DA REACÇÃO A sensibilidade da velocidade à variação da concentração de A e B Lei da velocidade. Imaginem uma reacção hipotética. Uma lei de velocidade genérica pode ser dada pela seguinte expressão onde K é a constante de velocidade e A e B as concentrações dos reagentes envolvidos nesta reacção. Note–se que as concentrações de A e B estão elevadas a um expoente x e y e que estes nada têm a ver com os coeficientes estequiométricos da reacção. Então nós podemos exprimir uma reacção hipotética por…. Nós sabemos que a velocidade de uma dada reacção resulta da variação da concentração de um dos seus componentes ao longo do tempo dA/dt. Por outro lado, esta variação ou velocidade vai depender da natureza dos componentes envolvidos Onde a velocidade da reacção pode ser pelo o produto das concentrações dos regentes envolvidos por uma constante Onde x e y são diferentes dos coeficientes estequiométricos. es Sendo que esta ordem só poderá ser determinada experimentalmente. Através da observação do impacto da concentração de A e B na velocidade de reacção .

8
Velocidade inicial (M/s)
Como Fazemos isto na prática? - Exemplo 2𝑁𝑂(𝑔)+2𝐻 2 𝑔 → 𝑁 2 𝑔 +2 𝐻 2 𝑂(𝑔) [NO] (M) [H2](M) Velocidade inicial (M/s) 5× 10 −3 2× 10 −3 1,25× 10 −5 10× 10 −3 5,00× 10 −5 4× 10 −3 10,00× 10 −5 Velocidade de reacção quando [NO]= 𝟏𝟐× 𝟏𝟎 −𝟑 M e [H2]= 𝟔× 𝟏𝟎 −𝟑 M A reacção do Oxido nítrico com o hidrogénio a 1280ºC Determinar: Lei da velocidade, Constante de velocidade
+2𝐻 2 𝑔 → 𝑁 2 𝑔 +2 𝐻 2 𝑂(𝑔) [NO] (M) [H2](M) Velocidade inicial (M/s) 5× 10 −3. 2× 10 −3. 1,25× 10 −5. 10× 10 −3. 5,00× 10 −5. 4× 10 −3. 10,00× 10 −5. Velocidade de reacção quando [NO]= 𝟏𝟐× 𝟏𝟎 −𝟑 M e [H2]= 𝟔× 𝟏𝟎 −𝟑 M. A reacção do Oxido nítrico com o hidrogénio a 1280ºC. Determinar: Lei da velocidade, Constante de velocidade.")
9
[NO] (M) [H2](M) Velocidade inicial (M/s) 5,00× 10 −3 2,00× 10 −3 1,25× 10 −5 10,00× 10 −3 5,00,× 10 −5 4,00× 10 −3 10,00× 10 −5 1 2 3 Como determinar x e y? Lei da velocidade 𝑽𝒆𝒍𝒐𝒄𝒊𝒅𝒂𝒅𝒆=𝜥 𝑨 𝒙 𝑩 𝒚 Mantendo a concentração de H2 constante e dobrando concentração de NO vemos que velocidade da reacção quadruplica 4=Κ 𝐴 𝑥 𝐵 𝑦 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 1 =Κ 𝐴 𝑥 𝐵 𝑦 𝐾= 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 𝐹 2 [𝐶𝑙 𝑂 2 ] = 1,2× 10 −3 M/s 0,10 𝑀 0,010 =1,2 𝑀𝑠 −1 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 2 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 1 = Κ 𝐴 𝑥 𝐵 𝑦 Κ 𝐴 𝑥 𝐵 𝑦 4= Κ 𝑁𝑂 𝑥 𝐻 2 𝑦 Κ 𝑁𝑂 𝑥 𝐻 2 𝑦 4= Κ 10× 10 −3 𝑥 2× 10 −3 𝑦 Κ 5× 10 −3 𝑥 2× 10 − 𝑦 4= 2 𝑥 𝑥=2 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=Κ 𝑁𝑂 𝐻 2 1 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 3 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 2 = Κ 𝐴 𝑥 𝐵 𝑦 Κ 𝐴 𝑥 𝐵 𝑦 2= Κ 𝑁𝑂 𝑥 𝐻 2 𝑦 Κ 𝑁𝑂 𝑥 𝐻 2 𝑦 A ordem global é dada por (2+1) ou seja a reacção é de terceira ordem 2= Κ 10× 10 −3 𝑥 4× 10 −3 𝑦 Κ 10× 10 −3 𝑥 2 × 10 − 𝑦 2= 2 𝑦 𝑦=1 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 2 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 1 = Κ 𝐴 𝑥 𝐵 𝑦 Κ 𝐴 𝑥 𝐵 𝑦
![[NO] (M) [H2](M) Velocidade inicial (M/s) 5,00× 10 −3. 2,00× 10 −3. 1,25× 10 −5. 10,00× 10 −3.](http://slideplayer.com.br/slide/12187690/71/images/9/%5BNO%5D+%28M%29+%5BH2%5D%28M%29+Velocidade+inicial+%28M%2Fs%29+5%2C00%C3%97+10+%E2%88%923.+2%2C00%C3%97+10+%E2%88%923.+1%2C25%C3%97+10+%E2%88%925.+10%2C00%C3%97+10+%E2%88%923..jpg "5,00,× 10 −5. 4,00× 10 −3. 10,00× 10 − Como determinar x e y Lei da velocidade. 𝑽𝒆𝒍𝒐𝒄𝒊𝒅𝒂𝒅𝒆=𝜥 𝑨 𝒙 𝑩 𝒚. Mantendo a concentração de H2 constante e dobrando concentração de NO vemos que velocidade da reacção quadruplica. 4=Κ 𝐴 𝑥 𝐵 𝑦 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 1 =Κ 𝐴 𝑥 𝐵 𝑦. 𝐾= 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 𝐹 2 [𝐶𝑙 𝑂 2 ] = 1,2× 10 −3 M/s 0,10 𝑀 0,010 =1,2 𝑀𝑠 −1. 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 2 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 1 = Κ 𝐴 𝑥 𝐵 𝑦 Κ 𝐴 𝑥 𝐵 𝑦. 4= Κ 𝑁𝑂 𝑥 𝐻 2 𝑦 Κ 𝑁𝑂 𝑥 𝐻 2 𝑦. 4= Κ 10× 10 −3 𝑥 2× 10 −3 𝑦 Κ 5× 10 −3 𝑥 2× 10 −3 2 𝑦. 4= 2 𝑥. 𝑥=2. 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=Κ 𝑁𝑂 2 𝐻 2 1. 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 3 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 2 = Κ 𝐴 𝑥 𝐵 𝑦 Κ 𝐴 𝑥 𝐵 𝑦. 2= Κ 𝑁𝑂 𝑥 𝐻 2 𝑦 Κ 𝑁𝑂 𝑥 𝐻 2 𝑦. A ordem global é dada por (2+1) ou seja a reacção é de terceira ordem. 2= Κ 10× 10 −3 𝑥 4× 10 −3 𝑦 Κ 10× 10 −3 𝑥 2 × 10 −3 2 𝑦. 2= 2 𝑦. 𝑦=1. 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 2 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 1 = Κ 𝐴 𝑥 𝐵 𝑦 Κ 𝐴 𝑥 𝐵 𝑦.")
10
Velocidade inicial (M/s)
[NO] (M) [H2](M) Velocidade inicial (M/s) 5,00× 10 −3 2,00× 10 −3 1,25× 10 −5 10,00× 10 −3 5,00,× 10 −5 4,00× 10 −3 10,00× 10 −5 A constante de velocidade k 𝑣=Κ 𝑁𝑂 𝐻 2 1 𝑽𝒆𝒍𝒐𝒄𝒊𝒅𝒂𝒅𝒆=𝜥 𝑨 𝒙 𝑩 𝒚 𝑘= 𝑣𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 𝑁𝑂 𝐻 2 1 𝑘= 1,25× 10 −5 5,00× 10 − ,00× 10 −3 1 𝑘= 2,50× 10 2 𝑀 −2 𝑠 −1 (c) Velocidade de reacção quando [NO]= 𝟏𝟐,𝟎𝟎× 𝟏𝟎 −𝟑 M e [H2]= 𝟔,𝟎𝟎× 𝟏𝟎 −𝟑 M 𝑽𝒆𝒍𝒐𝒄𝒊𝒅𝒂𝒅𝒆=2,50× 𝟏𝟐,𝟎𝟎× 𝟏𝟎 −𝟑 𝟐 𝟔,𝟎𝟎× 𝟏𝟎 −𝟑 𝟏 𝑽𝒆𝒍𝒐𝒄𝒊𝒅𝒂𝒅𝒆=2,20× 10 −4 𝑀 𝑠 −1
 [H2](M) Velocidade inicial (M/s) 5,00× 10 −3. 2,00× 10 −3. 1,25× 10 −5. 10,00× 10 −3. 5,00,× 10 −5. 4,00× 10 −3. 10,00× 10 −5. A constante de velocidade k. 𝑣=Κ 𝑁𝑂 2 𝐻 2 1. 𝑽𝒆𝒍𝒐𝒄𝒊𝒅𝒂𝒅𝒆=𝜥 𝑨 𝒙 𝑩 𝒚. 𝑘= 𝑣𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 𝑁𝑂 2 𝐻 2 1. 𝑘= 1,25× 10 −5 5,00× 10 −3 2 2,00× 10 −3 1. 𝑘= 2,50× 10 2 𝑀 −2 𝑠 −1. (c) Velocidade de reacção quando [NO]= 𝟏𝟐,𝟎𝟎× 𝟏𝟎 −𝟑 M e [H2]= 𝟔,𝟎𝟎× 𝟏𝟎 −𝟑 M. 𝑽𝒆𝒍𝒐𝒄𝒊𝒅𝒂𝒅𝒆=2,50× 10 2 𝟏𝟐,𝟎𝟎× 𝟏𝟎 −𝟑 𝟐 𝟔,𝟎𝟎× 𝟏𝟎 −𝟑 𝟏. 𝑽𝒆𝒍𝒐𝒄𝒊𝒅𝒂𝒅𝒆=2,20× 10 −4 𝑀 𝑠 −1.")
11
ln 𝐴 𝑡 =−𝑘𝑡+ ln [𝐴] 0 Relação entre a concentração e o tempo
“A lei da velocidade podem ainda ser utilizadas para determinar a concentração de reagentes em qualquer instante da reacção” 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=− Δ[𝐴] Δ𝑡 1 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=𝑘[𝐴] A Produto Reacção de 1ª Ordem 𝑘 𝐴 =− Δ[𝐴] Δ𝑡 𝑘 𝐴 =− 𝑑[𝐴] 𝑑𝑡 𝐴 𝑡 𝑡 ln 𝐴 𝑡 𝑡 −𝑘 0 𝑡 𝑑𝑡 = 𝐴 𝐴 𝑡 1 𝐴 𝑑[𝐴] 𝑘= 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 [𝐴] −𝑘𝑑𝑡= 𝑑[𝐴] 𝐴 𝑘=𝑚 −𝑘𝑡= ln [𝐴] 𝑡 − ln [𝐴] 0 ln 𝐴 𝑡 =−𝑘𝑡+ ln [𝐴] 0 𝑦=𝑚𝑥+b
![ln 𝐴 𝑡 =−𝑘𝑡+ ln [𝐴] 0 Relação entre a concentração e o tempo](http://slideplayer.com.br/slide/12187690/71/images/11/ln+%F0%9D%90%B4+%F0%9D%91%A1+%3D%E2%88%92%F0%9D%91%98%F0%9D%91%A1%2B+ln+%5B%F0%9D%90%B4%5D+0+Rela%C3%A7%C3%A3o+entre+a+concentra%C3%A7%C3%A3o+e+o+tempo.jpg "A lei da velocidade podem ainda ser utilizadas para determinar a concentração de reagentes em qualquer instante da reacção 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=− Δ[𝐴] Δ𝑡. 1. 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=𝑘[𝐴] A Produto. Reacção de 1ª Ordem. 𝑘 𝐴 =− Δ[𝐴] Δ𝑡. 𝑘 𝐴 =− 𝑑[𝐴] 𝑑𝑡. 𝐴 𝑡. 𝑡. ln 𝐴 𝑡. 𝑡. −𝑘 0 𝑡 𝑑𝑡 = 𝐴 0 𝐴 𝑡 1 𝐴 𝑑[𝐴] 𝑘= 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒 [𝐴] −𝑘𝑑𝑡= 𝑑[𝐴] 𝐴. 𝑘=𝑚. −𝑘𝑡= ln [𝐴] 𝑡 − ln [𝐴] 0. ln 𝐴 𝑡 =−𝑘𝑡+ ln [𝐴] 0. 𝑦=𝑚𝑥+b.")
12
Relação entre a concentração e o tempo
“À medida que a reacção avança, a concentração dos reagentes diminui” - um outra medida que relaciona a concentração com tempo – é o TEMPO DE MEIA-VIDA “ 𝒕 𝟏 𝟐 É o tempo necessário para a concentração do reagente diminuir para metade do seu valor inicial ln 𝐴 𝑡 =−𝑘𝑡+ ln [𝐴] 0 𝑡= 1 𝑘 ln [𝐴] 0 [𝐴] 𝑡 𝑡 = 1 𝑘 ln 2[𝐴] 0 [𝐴] 0 [𝐴] 𝑡 = [𝐴] 0 /2 𝑡 = 1 𝑘 ln 2[𝐴] 0 [𝐴] 0 𝑡 = 0,693 𝑘 Ln 2=0,693

13
Relação entre a concentração e o tempo
Reacções de segunda ordem – ”quando a velocidade depende da concentração de um reagente ao quadrado, ou quando a soma das concentrações (x+y) é 2. 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=− Δ[𝐴] Δ𝑡 A Produto 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=𝑘 [𝐴] 2 A+B Produto 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=𝑘[𝐴][B] Reacção de 2ª Ordem −𝑘 0 𝑡 𝑑𝑡 = 𝐴 𝐴 𝑡 [𝐴] −2 𝑑[𝐴] 𝑘 [𝐴] 2 =− 𝑑[𝐴] 𝑑𝑡 −𝑘𝑑𝑡= 𝑑[𝐴] [𝐴] 2 −𝑘𝑡= 𝐴 −1 𝑡 − 𝐴 −1 0 1 [𝐴] 𝑡 𝑡 1 [𝐴] 𝑡 =𝑘𝑡+ 1 [𝐴] 0
 é 2. 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=− Δ[𝐴] Δ𝑡. A Produto. 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=𝑘 [𝐴] 2. A+B Produto. 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=𝑘[𝐴][B] Reacção de 2ª Ordem. −𝑘 0 𝑡 𝑑𝑡 = 𝐴 0 𝐴 𝑡 [𝐴] −2 𝑑[𝐴] 𝑘 [𝐴] 2 =− 𝑑[𝐴] 𝑑𝑡. −𝑘𝑑𝑡= 𝑑[𝐴] [𝐴] 2. −𝑘𝑡= 𝐴 −1 𝑡 − 𝐴 − [𝐴] 𝑡. 𝑡. 1 [𝐴] 𝑡 =𝑘𝑡+ 1 [𝐴] 0.")
14
Relação entre a concentração e o tempo
TEMPO DE MEIA-VIDA para reacções de segunda ordem pode ser dado por: 1 [𝐴] 𝑡 =𝑘𝑡+ 1 [𝐴] 0 1 [𝐴] 0 /2 =𝑘 𝑡 [𝐴] 0 [𝐴] 𝑡 = [𝐴] 0 /2 TEMPO DE MEIA-VIDA de uma reacção de segunda ordem é inversamente proporcional à concentração inicial do reagente. 𝑡 = 1 𝑘 [𝐴] 0 Tempo de meia vida é mais baixo nos instantes iniciais Maior numero de moléculas disponíveis para colidirem umas com as outras

15
2ª Ordem 𝑑𝑥 𝑑𝑡 =𝑘 𝐴 [𝐵] 𝑑𝑥 𝑑𝑡 =𝑘 𝐴 0 −𝑥 𝐵 0 −𝑥 𝑑𝑥 𝐴 0 −𝑥 𝐵 0 −𝑥 =𝑘𝑑𝑡
𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=− 𝑑 𝐴 𝑑𝑡 =− 𝑑 𝐵 𝑑𝑡 =𝑘[𝐴][𝐵] A B Produtos 𝐴 = 𝐴 0 −𝑥 𝐴 0 𝐵 0 𝐵 = 𝐵 0 −𝑥 𝐴 0 −𝑥 𝐵 0 −𝑥 𝑥 𝑑𝑥 𝑑𝑡 =𝑘 𝐴 [𝐵] 𝑑𝑥 𝑑𝑡 =𝑘 𝐴 0 −𝑥 𝐵 0 −𝑥 𝑑𝑥 𝐴 0 −𝑥 𝐵 0 −𝑥 =𝑘𝑑𝑡 1 [𝐵] 0 − 𝐴 ln [ 𝐴 −𝑥] [𝐵] 0 [ 𝐵 0 −𝑥][ 𝐴] 0 =𝑘𝑡
![2ª Ordem 𝑑𝑥 𝑑𝑡 =𝑘 𝐴 [𝐵] 𝑑𝑥 𝑑𝑡 =𝑘 𝐴 0 −𝑥 𝐵 0 −𝑥 𝑑𝑥 𝐴 0 −𝑥 𝐵 0 −𝑥 =𝑘𝑑𝑡](http://slideplayer.com.br/slide/12187690/71/images/15/2%C2%AA+Ordem+%F0%9D%91%91%F0%9D%91%A5+%F0%9D%91%91%F0%9D%91%A1+%3D%F0%9D%91%98+%F0%9D%90%B4+%5B%F0%9D%90%B5%5D+%F0%9D%91%91%F0%9D%91%A5+%F0%9D%91%91%F0%9D%91%A1+%3D%F0%9D%91%98+%F0%9D%90%B4+0+%E2%88%92%F0%9D%91%A5+%F0%9D%90%B5+0+%E2%88%92%F0%9D%91%A5+%F0%9D%91%91%F0%9D%91%A5+%F0%9D%90%B4+0+%E2%88%92%F0%9D%91%A5+%F0%9D%90%B5+0+%E2%88%92%F0%9D%91%A5+%3D%F0%9D%91%98%F0%9D%91%91%F0%9D%91%A1.jpg "𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒=− 𝑑 𝐴 𝑑𝑡 =− 𝑑 𝐵 𝑑𝑡 =𝑘[𝐴][𝐵] A + B Produtos. 𝐴 = 𝐴 0 −𝑥. 𝐴 0. 𝐵 0. 𝐵 = 𝐵 0 −𝑥. 𝐴 0 −𝑥. 𝐵 0 −𝑥. 𝑥. 𝑑𝑥 𝑑𝑡 =𝑘 𝐴 [𝐵] 𝑑𝑥 𝑑𝑡 =𝑘 𝐴 0 −𝑥 𝐵 0 −𝑥. 𝑑𝑥 𝐴 0 −𝑥 𝐵 0 −𝑥 =𝑘𝑑𝑡. 1 [𝐵] 0 − 𝐴 0 ln [ 𝐴 −𝑥] [𝐵] 0 [ 𝐵 0 −𝑥][ 𝐴] 0 =𝑘𝑡.")
16
As reacções químicas ocorrem como consequência de colisões entre as moléculas dos reagentes.
Aplicando a teoria das colisões à cinética química. Então deveremos esperar que a velocidade da reacção seja directamente proporcional ao número de colisões moleculares por unidade de tempo… 𝑉𝑒𝑙𝑜𝑐𝑖𝑑𝑎𝑑𝑒∝ 𝑛𝑢𝑚𝑒𝑟𝑜 𝑑𝑒 𝑐𝑜𝑙𝑖𝑠õ𝑒𝑠 𝑡 Será mesmo assim?

17
Energia de activação e dependência das constantes de velocidade relativamente á temperatura
Quando duas moléculas colidem parte da energia cinética é convertida e energia vibracional Se a Energia cinética for suficientemente elevada quando as moléculas colidirem então a energia vibracional resultante será suficiente para quebrar algumas ligações químicas.

18
Energia necessária para que se inicie a reacção química
Energia de activação e dependência das constantes de velocidade relativamente á temperatura Quando duas moléculas colidem têm de possuir uma energia cinética igual ou superior à sua Energia de Activação ( 𝑬 𝒂 ). Energia necessária para que se inicie a reacção química Esta Energia mínima é fundamental para que as moléculas formem um complexo activado - Estado de Transição.
. Energia necessária para que se inicie a reacção química. Esta Energia mínima é fundamental para que as moléculas formem um complexo activado - Estado de Transição.")
19
calor Energia de activação e dependência das constantes de velocidade relativamente á temperatura Esta reacção será acompanhada da libertação de calor se os produtos forem mais estáveis que os reagentes - Reacção exotérmica Caso contrário irão absorver calor do meio circundante - Reacção endotérmica

20
Energia de activação e dependdencia das consatntes de velocidade relativamente á temperatura
Estado de transição Estado de transição Estado de transição 𝑬 𝒂 𝑬 𝒂 Energia Potencial A+B C+D C+D A+B Progresso da reacção

21
A dependência da constante de velocidade de uma reacção, relativamente à temperatura pode ser expressa pela - Equação de Arrehenius 𝑘=𝐴 𝑒 − 𝐸 𝑎 𝑅𝑇 Chama-se factor de frequência à quantidade A, que representa a frequência das colisões – Constante - para um dado sistema reaccional numa gama de temperaturas bastante alargada.

22
Aumento da Temperatura conduz a um Aumento da velocidade
Que informação nos dá a constante de Equação de Arrehenius? ln 𝑘 = ln 𝐴 𝑒 − 𝐸 𝑎 𝑅𝑇 𝑘=𝐴 𝑒 − 𝐸 𝑎 𝑅𝑇 Aumento da energia de activação diminuição da constante de velocidade (k). Aumento da Temperatura conduz a um Aumento da velocidade Sinal negativo associado ao exponente implica:
. Aumento da Temperatura conduz a um Aumento da velocidade. Sinal negativo associado ao exponente implica:")
23
Como poderemos estimar a Energia de Activação para uma dada temperatura?
𝑘=𝐴 𝑒 − 𝐸 𝑎 𝑅𝑇 ln 𝑘 = ln 𝐴 𝑒 − 𝐸 𝑎 𝑅𝑇 ln 𝑘 = ln 𝐴 − 𝐸 𝑎 𝑅𝑇 ln 𝑘 =− 𝐸 𝑎 𝑅𝑇 + ln 𝐴 ln 𝑘 1 𝑇 𝑦=𝑚𝑥+b

24
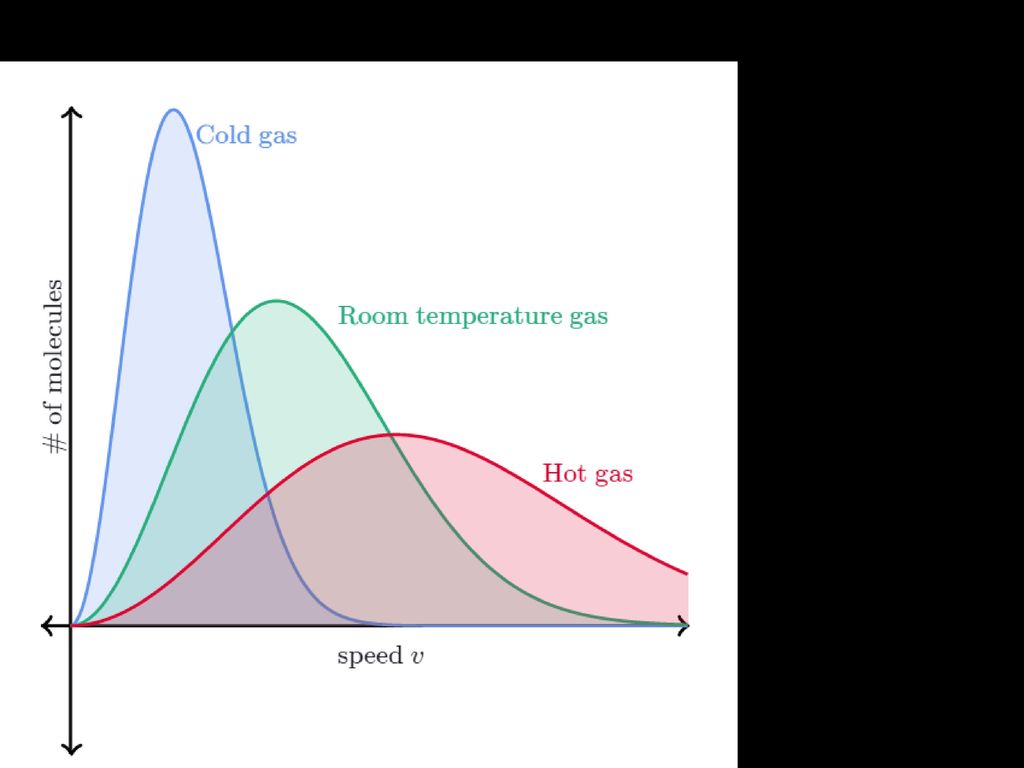
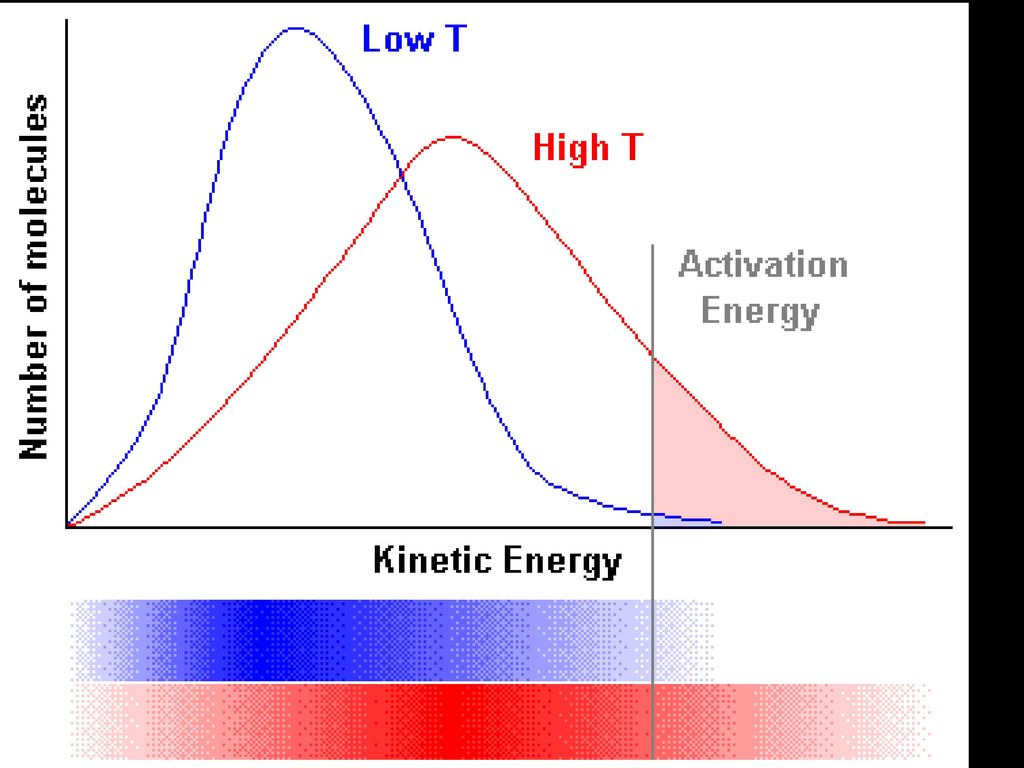
Será possível determinar a velocidade de colisão?
Distribuição de Maxwell -Boltzmann Área compreendida pela curva representa o número de partículas existentes no sistema Número de partículas Energia de Activação - Ea Energia mais provável energia Energia média

25
Distribuição de Maxwell -Boltzman
Aumento da temperatura o que acontece? A Área não varia pois o número de partículas não foi alterado Número de partículas Energia mais provável energia O número de partículas com mais energia aumentou Energia de Activação permanece inalterada, no entanto o número de partículas com energia superior à energia de activação subiu com o aumento da temperatura Energia média Maior velocidade pois as partículas estão mais energéticas, mais rápidas e colidem mais facilmente

26
Distribuição de Maxwell -Boltzman
Resumindo Quando T aumenta: A energia mais provável de partículas na amostra também aumenta. O número de partículas com a energia mais provável diminui. A energia média das partículas na amostra aumenta. Número de partículas com a energia média diminui. Área sob a curva de Maxwell –Boltzmann. permanece igual. A Energia de Activação permanece igual. O número de partículas na amostra que contém. Uma energia superior à energia de activação aumenta.

27
Mas com que velocidade é realizada esta colisão?
Distribuição de Maxwell -Boltzmann

28
velocidade mais provável
𝑓 𝑥 =4𝜋 𝑚 2𝜋 𝐾 𝐵 𝑇 𝑣 2 𝑒 −𝑚 𝑣 2 2 𝐾 𝐵 𝑇 Número de partículas 𝑓 𝑥 = 𝐶 1 𝑣 2 𝑒 − 𝐶 2 𝑣 2 𝑓 ′ 𝑥 = 𝐶 1 𝑣 2 𝑒 −𝐶 2 𝑣 2 𝑓 𝑥 𝑔 ′ 𝑥 +𝑔 𝑥 𝑓 ′ 𝑥 =0 velocidade mais provável velocidade 𝑓 ′ 𝑥 =0 𝐶 1 𝑣 2 −2 𝐶 2 𝑣 𝑒 − 𝐶 2 𝑣 2 +2𝑣 𝐶 1 𝑒 −𝐶 2 𝑣 2 =0 −2 𝐶 2 𝐶 1 𝑣 3 𝑒 −𝐶 2 𝑣 2 +2𝑣 𝐶 1 𝑒 − 𝐶 2 𝑣 2 =0 Máximo desta função −𝑣 2 𝐶 2 +1=0 2𝑣𝐶 1 𝑒 −𝐶 2 𝑣 2 ( −𝑣 2 𝐶 2 +1)=0 𝑣= 1 𝐶 2 𝑣= 1 𝑚 2 𝐾 𝐵 𝑇 𝑣 2 = 1 𝐶 2 𝑣 𝑀𝑃 = 2 𝐾 𝐵 𝑇 𝑚 𝑣 𝑀𝑃 = 2𝑅𝑇 𝑀
=0. 𝑣= 1 𝐶 2. 𝑣= 1 𝑚 2 𝐾 𝐵 𝑇. 𝑣 2 = 1 𝐶 2. 𝑣 𝑀𝑃 = 2 𝐾 𝐵 𝑇 𝑚. 𝑣 𝑀𝑃 = 2𝑅𝑇 𝑀.")
29
𝑣∗𝑓 𝑣 =𝑣∗4𝜋 𝑚 2𝜋 𝐾 𝐵 𝑇 3 2 𝑣 2 𝑒 −𝑚 𝑣 2 2 𝐾 𝐵 𝑇
O que faríamos se quiséssemos saber quantas partículas estavam presentes numa dada região da distribuição? 𝑣 1 𝑣 2 𝑓(𝑣) 𝑑𝑣 0 ∞ 𝑓(𝑣) 𝑑𝑣 0 ∞ 𝑣×𝑓(𝑣) 𝑑𝑣= 𝑉 𝑀𝐷 𝑣∗𝑓 𝑣 =𝑣∗4𝜋 𝑚 2𝜋 𝐾 𝐵 𝑇 𝑣 2 𝑒 −𝑚 𝑣 2 2 𝐾 𝐵 𝑇 Número de partículas 𝑣𝑓 𝑣 = 𝐶 1 𝑣 3 𝑒 − 𝐶 2 𝑣 2 velocidade 0 ∞ 𝐶 1 𝑣 3 𝑒 − 𝐶 2 𝑣 2 𝑑𝑣= 𝑉 𝑀𝐷 Velocidade média 𝑣 1 𝑣 2 𝐶 1 0 ∞ 𝑣 3 𝑒 − 𝐶 2 𝑣 2 𝑑𝑣= 𝑉 𝑀𝐷 𝐶 1 1! 2 𝐶 𝐶 𝐶 = 𝑉 𝑀𝐷 0 ∞ 𝑥 2𝑛+1 𝑒 −𝑎 𝑥 2 𝑑𝑥= 𝑛! 2 𝑎 𝑛+1
 𝑑𝑣. 0 ∞ 𝑓(𝑣) 𝑑𝑣. 0 ∞ 𝑣×𝑓(𝑣) 𝑑𝑣= 𝑉 𝑀𝐷. 𝑣∗𝑓 𝑣 =𝑣∗4𝜋 𝑚 2𝜋 𝐾 𝐵 𝑇 3 2 𝑣 2 𝑒 −𝑚 𝑣 2 2 𝐾 𝐵 𝑇. Número de partículas. 𝑣𝑓 𝑣 = 𝐶 1 𝑣 3 𝑒 − 𝐶 2 𝑣 2. velocidade. 0 ∞ 𝐶 1 𝑣 3 𝑒 − 𝐶 2 𝑣 2 𝑑𝑣= 𝑉 𝑀𝐷. Velocidade média. 𝑣 1. 𝑣 2. 𝐶 1 0 ∞ 𝑣 3 𝑒 − 𝐶 2 𝑣 2 𝑑𝑣= 𝑉 𝑀𝐷. 𝐶 1 1! 2 𝐶 𝐶 1 2 𝐶 2 2 = 𝑉 𝑀𝐷. 0 ∞ 𝑥 2𝑛+1 𝑒 −𝑎 𝑥 2 𝑑𝑥= 𝑛! 2 𝑎 𝑛+1.")
30
𝑣∗𝑓 𝑣 =𝑣∗4𝜋 𝑚 2𝜋 𝐾 𝐵 𝑇 3 2 𝑣 2 𝑒 −𝑚 𝑣 2 2 𝐾 𝐵 𝑇 𝐶 1 2 𝐶 2 2 = 𝑉 𝑀𝐷
𝑣∗𝑓 𝑣 =𝑣∗4𝜋 𝑚 2𝜋 𝐾 𝐵 𝑇 𝑣 2 𝑒 −𝑚 𝑣 2 2 𝐾 𝐵 𝑇 𝐶 𝐶 = 𝑉 𝑀𝐷 4𝜋 𝑚 2𝜋 𝐾 𝐵 𝑇 𝑚 2 𝐾 𝐵 𝑇 2 = 𝑉 𝑀𝐷 2 2𝜋 𝑚 2𝜋 𝐾 𝐵 𝑇 × 1 𝑚 2 𝐾 𝐵 𝑇 2 = 𝑉 𝑀𝐷 2𝜋 𝑚 2𝜋 𝐾 𝐵 𝑇 × 2 𝐾 𝐵 𝑇 𝑚 −2 = 𝑉 𝑀𝐷 2 1 𝜋 − 𝜋 𝑚 2 𝐾 𝐵 𝑇 × 𝑚 2 𝐾 𝐵 𝑇 −2 = 𝑉 𝑀𝐷 ∗2 ∗2

31
velocidade mais provável
2 1 𝜋 − 𝜋 𝑚 2 𝐾 𝐵 𝑇 × 𝑚 2 𝐾 𝐵 𝑇 − 4 2 = 𝑉 𝑀𝐷 2 1 𝜋 𝑚 2 𝐾 𝐵 𝑇 − 1 2 = 𝑉 𝑀𝐷 2 1 𝜋 𝐾 𝐵 𝑇 𝑚 = 𝑉 𝑀𝐷 velocidade mais provável velocidade média 2 2 𝐾 𝐵 𝑇 𝜋𝑚 = 𝑉 𝑀𝐷 8 𝐾 𝐵 𝑇 𝜋𝑚 = 𝑉 𝑀𝐷 8𝑅𝑇 𝜋𝑀 = 𝑉 𝑀𝐷 𝑣 𝑀𝑃 = 2𝑅𝑇 𝑀

34
Aula 3 - Cinética Química
QFL 0464

35
Velocidade de formação de 𝐵= 𝐾 𝑟 𝐴
Estado de transição Sentido direto AB Sentido reverso A B Estado de transição 𝐵= [𝐵] 𝑒𝑞 [𝐴] 𝑒𝑞 = 𝐾 𝑟 𝐾 𝑟 ′ Velocidade de formação de 𝐵= 𝐾 𝑟 𝐴 𝑬 𝒂 Velocidade de de composição de 𝐵= 𝐾 𝑟 ′ 𝐵 𝑬 𝒂 Velocidade de formação de 𝐵= 𝐾 𝑟 𝐴 A 𝑑[𝐵] 𝑑𝑡 = 𝐾 𝑟 𝐴 − 𝐾 𝑟 ′ [B] Velocidade global do processo em relação a B 𝐵= 𝐾 𝑟 ′ 𝐵 Velocidade global do processo em relação a B B 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑟 𝐴 + 𝐾 𝑟 ′ [B] Progresso da reação 𝐾= [𝐵] 𝑒𝑞 [𝐴] 𝑒𝑞 = 𝐾 𝑟 𝐾 𝑟 ′ A relação entre as constantes cinéticas da reação direta e inversa Ajuda-nos a estabelecer uma relação entre a cinética e o equilíbrio químico

36
E quando ainda não estamos no Equilíbrio?
𝐾= [𝐵] 𝑒𝑞 [𝐴] 𝑒𝑞 = 𝐾 𝑟 𝐾 𝑟 ′ Equilíbrio 1 𝑎𝑥+𝑏 𝑑𝑥=𝑑𝑡 1 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 𝑑[𝐴]=𝑑𝑡 E quando ainda não estamos no Equilíbrio? [𝐴] 0 = 𝐵 + 𝐴 𝐵 = [𝐴] 0 − 𝐴 1 𝑎𝑥+𝑏 𝑑[𝑥]= ln (𝑎𝑥+𝑏) 𝑎 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑟 𝐴 + 𝐾 𝑟 ′ [B] 𝑓 𝑥 =𝑓 𝐴 −𝑓( 𝐴 0 ) 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑟 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 − 𝐴 [𝐴] 0 [𝐴] 1 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 𝑑[𝐴]= 0 𝑡 𝑑𝑡 𝑑[𝐴] 𝑑𝑡 =− 𝐴 𝐾 𝑟 + 𝐾 𝑟 ′ + 𝐾 𝑟 ′ [𝐴] 0 𝑑[𝐴] − 𝐴 𝐾 𝑟 + 𝐾 𝑟 ′ + 𝐾 𝑟 ′ [𝐴] 0 =𝑑𝑡 𝑎=− 𝐾 𝑟 + 𝐾 𝑟 ′ b= 𝐾 𝑟 ′ [𝐴] 0 1 𝑎𝑥+𝑏 𝑑[𝑥]= ln (𝑎𝑥+𝑏) 𝑎 Equação diferencial Integral
 𝑎. 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑟 𝐴 + 𝐾 𝑟 ′ [B] 𝑓 𝑥 =𝑓 𝐴 −𝑓( 𝐴 0 ) 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑟 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 − 𝐴. [𝐴] 0 [𝐴] 1 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 𝑑[𝐴]= 0 𝑡 𝑑𝑡. 𝑑[𝐴] 𝑑𝑡 =− 𝐴 𝐾 𝑟 + 𝐾 𝑟 ′ + 𝐾 𝑟 ′ [𝐴] 0. 𝑑[𝐴] − 𝐴 𝐾 𝑟 + 𝐾 𝑟 ′ + 𝐾 𝑟 ′ [𝐴] 0 =𝑑𝑡. 𝑎=− 𝐾 𝑟 + 𝐾 𝑟 ′ b= 𝐾 𝑟 ′ [𝐴] 0. 1 𝑎𝑥+𝑏 𝑑[𝑥]= ln (𝑎𝑥+𝑏) 𝑎. Equação diferencial. Integral.")
37
[𝐴] 0 [𝐴] 1 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 𝑑[𝐴]= 0 𝑡 𝑑𝑡
1 𝑎𝑥+𝑏 𝑑[𝑥]= ln (𝑎𝑥+𝑏) 𝑎 [𝐴] 0 [𝐴] 1 𝑎𝑥+𝑏 𝑑 𝑥 = ln 𝑎 𝐴 +𝑏 𝑎 − ln 𝑎 𝐴 0 +𝑏 𝑎 =𝑡 ln 𝑎 𝐴 +𝑏 𝑎 𝐴 0 +𝑏 =𝑎𝑡 𝑎=− 𝐾 𝑟 + 𝐾 𝑟 ′ b= 𝐾 𝑟 ′ [𝐴] 0 ln − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 0 + 𝐾 𝑟 ′ [𝐴] 0 =− 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 ln 𝑥 =𝑡→𝑥= 𝑒 𝑡 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 0 + 𝐾 𝑟 ′ [𝐴] 0 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡
![[𝐴] 0 [𝐴] 1 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 𝑑[𝐴]= 0 𝑡 𝑑𝑡](http://slideplayer.com.br/slide/12187690/71/images/37/%5B%F0%9D%90%B4%5D+0+%5B%F0%9D%90%B4%5D+1+%E2%88%92+%F0%9D%90%BE+%F0%9D%91%9F+%2B+%F0%9D%90%BE+%F0%9D%91%9F+%E2%80%B2+%F0%9D%90%B4+%2B+%F0%9D%90%BE+%F0%9D%91%9F+%E2%80%B2+%5B%F0%9D%90%B4%5D+0+%F0%9D%91%91%5B%F0%9D%90%B4%5D%3D+0+%F0%9D%91%A1+%F0%9D%91%91%F0%9D%91%A1.jpg "1 𝑎𝑥+𝑏 𝑑[𝑥]= ln (𝑎𝑥+𝑏) 𝑎. [𝐴] 0 [𝐴] 1 𝑎𝑥+𝑏 𝑑 𝑥 = ln 𝑎 𝐴 +𝑏 𝑎 − ln 𝑎 𝐴 0 +𝑏 𝑎 =𝑡. ln 𝑎 𝐴 +𝑏 𝑎 𝐴 0 +𝑏 =𝑎𝑡. 𝑎=− 𝐾 𝑟 + 𝐾 𝑟 ′ b= 𝐾 𝑟 ′ [𝐴] 0. ln − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 0 + 𝐾 𝑟 ′ [𝐴] 0 =− 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡. ln 𝑥 =𝑡→𝑥= 𝑒 𝑡. − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 0 + 𝐾 𝑟 ′ [𝐴] 0 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡.")
38
− 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 0 + 𝐾 𝑟 ′ [𝐴] 0 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡
− 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 − 𝐾 𝑟 𝐴 0 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 − 𝐾 𝑟 𝐴 𝐾 𝑟 ′ [𝐴] 0 𝐾 𝑟 𝐴 0 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 𝐾 𝑟 𝐴 0 − 𝐾 𝑟 ′ 𝐾 𝑟 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 𝐾 𝑟 𝐴 0 − 𝐾 𝑟 ′ 𝐾 𝑟 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 𝐴 𝐴 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐾 𝑟 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 + 𝐾 𝑟 ′ 𝐾 𝑟 𝐴 𝐴 0 = 𝐾 𝑟 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 + 𝐾 𝑟 ′ 𝐾 𝑟 𝐴 = 𝐾 𝑟 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 + 𝐾 𝑟 ′ 𝐾 𝑟 𝐴 0
![− 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 0 + 𝐾 𝑟 ′ [𝐴] 0 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡](http://slideplayer.com.br/slide/12187690/71/images/38/%E2%88%92+%F0%9D%90%BE+%F0%9D%91%9F+%2B+%F0%9D%90%BE+%F0%9D%91%9F+%E2%80%B2+%F0%9D%90%B4+%2B+%F0%9D%90%BE+%F0%9D%91%9F+%E2%80%B2+%5B%F0%9D%90%B4%5D+0+%E2%88%92+%F0%9D%90%BE+%F0%9D%91%9F+%2B+%F0%9D%90%BE+%F0%9D%91%9F+%E2%80%B2+%F0%9D%90%B4+0+%2B+%F0%9D%90%BE+%F0%9D%91%9F+%E2%80%B2+%5B%F0%9D%90%B4%5D+0+%3D+%F0%9D%91%92+%E2%88%92+%F0%9D%90%BE+%F0%9D%91%9F+%2B+%F0%9D%90%BE+%F0%9D%91%9F+%E2%80%B2+%F0%9D%91%A1.jpg "− 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 + 𝐾 𝑟 ′ [𝐴] 0 − 𝐾 𝑟 𝐴 0 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡. − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 − 𝐾 𝑟 𝐴 0 + 𝐾 𝑟 ′ [𝐴] 0 𝐾 𝑟 𝐴 0 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡. 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 𝐾 𝑟 𝐴 0 − 𝐾 𝑟 ′ 𝐾 𝑟 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡. 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐴 𝐾 𝑟 𝐴 0 − 𝐾 𝑟 ′ 𝐾 𝑟 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡. 𝐴 𝐴 0 𝐾 𝑟 + 𝐾 𝑟 ′ 𝐾 𝑟 = 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 + 𝐾 𝑟 ′ 𝐾 𝑟. 𝐴 𝐴 0 = 𝐾 𝑟 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 + 𝐾 𝑟 ′ 𝐾 𝑟. 𝐴 = 𝐾 𝑟 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑒 − 𝐾 𝑟 + 𝐾 𝑟 ′ 𝑡 + 𝐾 𝑟 ′ 𝐾 𝑟 𝐴 0.")
39
Tempo de relaxação A maioria das reações (e.g. Enzimáticas) ocorrem na escala de tempo dos ms e por isso podemos usar técnicas simples para avaliar - denominadas de ¨stop flow¨. No entanto, existem processos (e.g. formação de intermediários reacionais) cuja cinética ocorre a baixo da escala dos ms. – para estes casos temos que utilizar outro tipo de técnicas – técnicas de relaxação. Neste caso o termo relaxação significa o retorno ao equilíbrio. E este regresso ao equilíbrio é medido após uma mudança súbita das condições – exemplo pressão ou temperatura. Por exemplo um salto de temperatura de 10 K pode ser atingido em 1 μs O aumento de temperatura induzido por lazer é bastante utilizado para estudar a desnaturação de proteínas
 ocorrem na escala de tempo dos ms e por isso podemos usar técnicas simples para avaliar - denominadas de ¨stop flow¨. No entanto, existem processos (e.g. formação de intermediários reacionais) cuja cinética ocorre a baixo da escala dos ms. – para estes casos temos que utilizar outro tipo de técnicas – técnicas de relaxação. Neste caso o termo relaxação significa o retorno ao equilíbrio. E este regresso ao equilíbrio é medido após uma mudança súbita das condições – exemplo pressão ou temperatura. Por exemplo um salto de temperatura de 10 K pode ser atingido em 1 μs. O aumento de temperatura induzido por lazer é bastante utilizado para estudar a desnaturação de proteínas.")
40
Tempo de relaxação 𝐴 𝑒𝑞2 𝐴 𝑒𝑞1 𝐴 𝑒𝑞2 < 𝐴 𝑒𝑞1 Reação A B
𝐴 𝑒𝑞2 𝐴 𝑒𝑞1 𝐴 𝑒𝑞2 < 𝐴 𝑒𝑞1 Reação A B Antes do salto de temperatura nós temos: um equilibro entre A e B que segue as seguintes condições: 𝑣= 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑎1 𝐴 + 𝐾 𝑏1 [𝐵] Eq. em T1 relaxação Eq. em T2 [A] 𝐴 𝑒𝑞1 𝐴 𝑒𝑞2 +𝑥 𝐴 𝑒𝑞2 [B] 𝐵 𝑒𝑞1 𝐵 𝑒𝑞2 −𝑥 𝐵 𝑒𝑞2 𝑛𝑜 𝑒𝑞𝑢𝑖𝑙𝑘𝑖𝑏𝑟𝑖𝑜 𝑡𝑒𝑚𝑜𝑠 : 𝑑[𝐴] 𝑑𝑡 =0 E𝑛𝑡ã𝑜 𝑝𝑜𝑑𝑒𝑚𝑜𝑠 𝑒𝑠𝑐𝑟𝑒𝑣𝑒𝑟 : 𝐾 𝑎1 𝐴 𝑒𝑞1 = 𝐾 𝑏1 𝐵 𝑒𝑞1 𝐾 𝑎2 𝐴 𝑒𝑞2 = 𝐾 𝑏2 𝐵 𝑒𝑞2

41
Eq. em T1 relaxação Eq. em T2 [A] 𝐴 𝑒𝑞1 𝐴 𝑒𝑞2 +𝑥 𝐴 𝑒𝑞2 [B] 𝐵 𝑒𝑞1 𝐵 𝑒𝑞2 −𝑥 𝐵 𝑒𝑞2 𝐴 𝑒𝑞2 𝐴 𝑒𝑞1 𝐴 𝑒𝑞2 < 𝐴 𝑒𝑞1 𝐴 = 𝐴 𝑒𝑞2 +𝑥 𝐴 𝑒𝑞2 + 𝑥 ´ 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑎2 𝐴 𝑒𝑞2 +𝑥 + 𝐾 𝑏2 𝐵 𝑒𝑞2 −𝑥 ] . 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑎2 𝑥− 𝐾 𝑎2 𝐴 𝑒𝑞2 + 𝐾 𝑏2 𝐵 𝑒𝑞2 − 𝐾 𝑏2 𝑥 Como 𝐾 𝑎2 𝐴 𝑒𝑞2 = 𝐾 𝑏2 𝐵 𝑒𝑞2 então: 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑎2 𝑥− 𝐾 𝑏2 𝐵 𝑒𝑞2 + 𝐾 𝑏2 𝐵 𝑒𝑞2 − 𝐾 𝑏2 𝑥 e 𝐴 𝑒𝑞2 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑎2 𝑥− 𝐾 𝑏2 𝑥 𝑑[𝐴] 𝑑𝑡 =−𝑥 𝐾 𝑎2 + 𝐾 𝑏2 𝑑[𝐴] 𝑑𝑡 O que é [A] ? É 𝐴 𝑒𝑞2 +x O que é 𝑑 𝐴 ? 𝑑[𝐴] 𝑑𝑡 = 𝑑𝑥 𝑑𝑡 𝑑𝑥 𝑑𝑡 =−𝑥 𝐾 𝑎2 + 𝐾 𝑏2 Então podemos definir Isto é,
![Eq. em T1 relaxação. Eq. em T2. [A] 𝐴 𝑒𝑞1. 𝐴 𝑒𝑞2 +𝑥. 𝐴 𝑒𝑞2. [B] 𝐵 𝑒𝑞1. 𝐵 𝑒𝑞2 −𝑥. 𝐵 𝑒𝑞2.](http://slideplayer.com.br/slide/12187690/71/images/41/Eq.+em+T1+relaxa%C3%A7%C3%A3o.+Eq.+em+T2.+%5BA%5D+%F0%9D%90%B4+%F0%9D%91%92%F0%9D%91%9E1.+%F0%9D%90%B4+%F0%9D%91%92%F0%9D%91%9E2+%2B%F0%9D%91%A5.+%F0%9D%90%B4+%F0%9D%91%92%F0%9D%91%9E2.+%5BB%5D+%F0%9D%90%B5+%F0%9D%91%92%F0%9D%91%9E1.+%F0%9D%90%B5+%F0%9D%91%92%F0%9D%91%9E2+%E2%88%92%F0%9D%91%A5.+%F0%9D%90%B5+%F0%9D%91%92%F0%9D%91%9E2..jpg "𝐴 𝑒𝑞2. 𝐴 𝑒𝑞1. 𝐴 𝑒𝑞2 < 𝐴 𝑒𝑞1. 𝐴. = 𝐴 𝑒𝑞2 +𝑥. 𝐴 𝑒𝑞2 + 𝑥 ´ 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑎2 𝐴 𝑒𝑞2 +𝑥 + 𝐾 𝑏2 𝐵 𝑒𝑞2 −𝑥 ] . 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑎2 𝑥− 𝐾 𝑎2 𝐴 𝑒𝑞2 + 𝐾 𝑏2 𝐵 𝑒𝑞2 − 𝐾 𝑏2 𝑥. Como 𝐾 𝑎2 𝐴 𝑒𝑞2 = 𝐾 𝑏2 𝐵 𝑒𝑞2 então: 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑎2 𝑥− 𝐾 𝑏2 𝐵 𝑒𝑞2 + 𝐾 𝑏2 𝐵 𝑒𝑞2 − 𝐾 𝑏2 𝑥. e 𝐴 𝑒𝑞2. 𝑑[𝐴] 𝑑𝑡 =− 𝐾 𝑎2 𝑥− 𝐾 𝑏2 𝑥. 𝑑[𝐴] 𝑑𝑡 =−𝑥 𝐾 𝑎2 + 𝐾 𝑏2. 𝑑[𝐴] 𝑑𝑡. O que é [A] É 𝐴 𝑒𝑞2 +x. O que é 𝑑 𝐴 𝑑[𝐴] 𝑑𝑡 = 𝑑𝑥 𝑑𝑡. 𝑑𝑥 𝑑𝑡 =−𝑥 𝐾 𝑎2 + 𝐾 𝑏2. Então podemos definir. Isto é,")
42
Tempo que demora a acontecer todas estas variações ou seja o tempo de relaxação
𝐴 𝐴 𝑒𝑞2 𝑑𝑥 𝑑𝑡 =−𝑥 𝐾 𝑎2 + 𝐾 𝑏2 = 𝐴 𝑒𝑞2 +𝑥 𝐴 𝑒𝑞2 + 𝑥 ´ 𝑑𝑥 𝑥 =− 𝐾 𝑎2 + 𝐾 𝑏2 𝑑𝑡 . 𝑥 0 𝑥 1 𝑥 𝑑𝑥 =− 𝐾 𝑎2 + 𝐾 𝑏2 0 𝑡 𝑑𝑡 𝐴 𝑒𝑞1 ln 𝑥 𝑥 0 =− 𝐾 𝑎2 + 𝐾 𝑏2 𝑡 ln 𝑥 − ln 𝑥 0 =− 𝐾 𝑎2 + 𝐾 𝑏2 𝑡 𝑡 𝐾 𝑎2 + 𝐾 𝑏2 = 1 𝜏 𝑥= 𝑥 0 𝑒 −𝑡 𝐾 𝑎2 + 𝐾 𝑏2 𝑥= 𝑥 0 𝑒 − 1 𝜏

43
Reações consecutivas Neste caso os reagentes produzem um intermediário – que é uma espécie que não aprece no final da equação mas que é descrito no mecanismo. Por exemplo uma enzima de restrição corta DNA AI I P 𝑣 𝑑𝑒 𝑓𝑜𝑟𝑚𝑎çã𝑜 𝑑𝑒 𝐼= 𝐾 1 𝐴 A 𝑣 𝑑𝑒 𝑓𝑜𝑟𝑚𝑎çã𝑜 𝑑𝑒 𝑃= 𝐾 2 𝐴 ln [𝐴] =−𝑘𝑡+ ln [𝐴] 0 [𝐴]= [𝐴] 0 𝑒 −𝑘𝑡 I A velocidade de formação de I é a diferença entre taxa da sua taxa de formação e a taxa do seu consumo. P 𝐼 = 𝑑[𝐼] 𝑑𝑡 = 𝐾 1 𝐴 − 𝐾 2 [𝐼] 𝑑[𝐼] 𝑑𝑡 = 𝐾 1 [𝐴] 0 𝑒 −𝑘𝑡 − 𝐾 2 [𝐼] 𝐼 = 𝐾 1 𝐾 1 − 𝐾 2 𝑒 − 𝐾 1 𝑡 − 𝑒 − 𝐾 2 𝑡 [𝐴] 0

44
𝐼 = 𝐾 1 𝐾 1 − 𝐾 2 𝑒 − 𝐾 1 𝑡 − 𝑒 − 𝐾 2 𝑡 [𝐴] 0 𝑑[𝐼] 𝑑𝑡 =0 0= 𝐾 1 𝐾 1 − 𝐾 2 𝑒 − 𝐾 1 𝑡 − 𝑒 − 𝐾 2 𝑡 [𝐴] 0 0= 𝑒 − 𝐾 1 𝑡 − 𝑒 − 𝐾 2 𝑡 𝑒 − 𝐾 2 𝑡 = 𝑒 − 𝐾 1 𝑡 𝐾 2 𝑒 − 𝐾 2 𝑡 = 𝐾 1 𝑒 − 𝐾 1 𝑡 𝐾 2 𝐾 1 = 𝑒 − 𝐾 1 𝑡 𝑒 − 𝐾 2 𝑡 𝐾 2 𝐾 1 = 𝑒 𝑡( 𝐾 2 − 𝐾 1 ) ln 𝐾 2 𝐾 1 =𝑡( 𝐾 2 − 𝐾 1 ) 𝐾 2 𝑒 − 𝐾 2 𝑡 = 𝐾 1 𝑒 − 𝐾 1 𝑡 𝑡= 1 ( 𝐾 2 − 𝐾 1 ) ln 𝐾 2 𝐾 1 O tempo na máxima concentração ou o tempo que demora a atingir a máxima concentração
![𝐼 = 𝐾 1 𝐾 1 − 𝐾 2 𝑒 − 𝐾 1 𝑡 − 𝑒 − 𝐾 2 𝑡 [𝐴] 0 𝑑[𝐼] 𝑑𝑡 =0. 0= 𝐾 1 𝐾 1 − 𝐾 2 𝑒 − 𝐾 1 𝑡 − 𝑒 − 𝐾 2 𝑡 [𝐴] 0.](http://slideplayer.com.br/slide/12187690/71/images/44/%F0%9D%90%BC+%3D+%F0%9D%90%BE+1+%F0%9D%90%BE+1+%E2%88%92+%F0%9D%90%BE+2+%F0%9D%91%92+%E2%88%92+%F0%9D%90%BE+1+%F0%9D%91%A1+%E2%88%92+%F0%9D%91%92+%E2%88%92+%F0%9D%90%BE+2+%F0%9D%91%A1+%5B%F0%9D%90%B4%5D+0+%F0%9D%91%91%5B%F0%9D%90%BC%5D+%F0%9D%91%91%F0%9D%91%A1+%3D0.+0%3D+%F0%9D%90%BE+1+%F0%9D%90%BE+1+%E2%88%92+%F0%9D%90%BE+2+%F0%9D%91%92+%E2%88%92+%F0%9D%90%BE+1+%F0%9D%91%A1+%E2%88%92+%F0%9D%91%92+%E2%88%92+%F0%9D%90%BE+2+%F0%9D%91%A1+%5B%F0%9D%90%B4%5D+0..jpg "0= 𝑒 − 𝐾 1 𝑡 − 𝑒 − 𝐾 2 𝑡. 𝑒 − 𝐾 2 𝑡 = 𝑒 − 𝐾 1 𝑡. 𝐾 2 𝑒 − 𝐾 2 𝑡 = 𝐾 1 𝑒 − 𝐾 1 𝑡. 𝐾 2 𝐾 1 = 𝑒 − 𝐾 1 𝑡 𝑒 − 𝐾 2 𝑡. 𝐾 2 𝐾 1 = 𝑒 𝑡( 𝐾 2 − 𝐾 1 ) ln 𝐾 2 𝐾 1 =𝑡( 𝐾 2 − 𝐾 1 ) 𝐾 2 𝑒 − 𝐾 2 𝑡 = 𝐾 1 𝑒 − 𝐾 1 𝑡. 𝑡= 1 ( 𝐾 2 − 𝐾 1 ) ln 𝐾 2 𝐾 1. O tempo na máxima concentração ou o tempo que demora a atingir a máxima concentração.")
Apresentações semelhantes















 e/ou produto de sua decomposição é medida em.>")
 Defina sequências numéricas.>")











