Carregar apresentação
A apresentação está carregando. Por favor, espere

1
EIXO HIPOTÁLAMO - HIPÓFISE - GLANDULA ALVO

3
SECREÇÃO ADENOHIPOFISÁRIA - FEEDBACK NEGATIVO: a diminuição de um hormônio circulante estimula a secreção do hormônio hipofisário que controla a glândula que o produz - FEEDBACK POSITIVO: o aumento de um hormônio circulante estimula a secreção do hormônio hipofisário que controla a glândula que o produz

4
SISTEMAS DE FEEDBACK - LONGO: estabelecido entre hipotálamo/hipófise e
glandula alvo CURTO: estabelecido entre hipotálamo e hipófise ULTRACURTO: estabelecido no hipotálamo, onde a concentração de hormônios autorregula sua própria síntese e secreção

5
Antecedentes pessoais: catapora
CASO CLÍNICO RCL, 14 anos e 04 meses, sexo masculino, consulta por baixa estatura e atraso do desenvolvimento puberal. Mãe informa que o paciente nasceu de parto normal, gravidez sem intercorrências, e com 02 anos de idade começou a apresentar “crises de bronquite”, não tendo necessitado de internação em nenhum dos referidos episódios. Antecedentes pessoais: catapora Antecedentes familiares: mãe: Alt= 155 cm e menarca aos 12 anos; pai: Alt= 186 cm Altura alvo= cm Exame físico: Peso = 35,2 Kg; Alt= 137,5 cm; PA= 112 x 75 mmHg; FC= 76 bpm Exame físico sem particularidades, com estadio puberal I

6
Exames realizados: Idade óssea: compatível com 10 anos RX de crânio: calcificação em região supra-selar CT de crânio e sela túrcica: calcificação em região supra-selar, invadindo a região da sela túrcica Hemograma: normal; Urina I: normal; Protoparasitológico: negativo T4 livre= 0,98 ng% (0,7 - 2,0 ng%) TSH= 2,14 mUI/ml (0,5 – 4,5mUI/ml) Cortisol urinário de 24 horas: normal Megateste: Tempo Glicemia(mg%) PRL (ng/ml) LH (mU/ml) FSH (mU/ml) GH (ng/ml) 92 3,2 1,3 1,6 0,3 15 37 7,4 1,5 1,8 0,5 30 42 6,4 1,0 1,9 0,6 45 73 3,4 1,7 0,2 60 88 4,5 0.07
 TSH= 2,14 mUI/ml (0,5 – 4,5mUI/ml) Cortisol urinário de 24 horas: normal. Megateste: Tempo. Glicemia(mg%) PRL (ng/ml) LH (mU/ml) FSH (mU/ml) GH (ng/ml) 92. 3,2. 1,3. 1,6. 0, ,4. 1,5. 1,8. 0, ,4. 1,0. 1,9. 0, ,4. 1,7. 0, ,")
7
Evolução: Craniotomia transfrontal para retirada da tumoração calcificada Diabetes insípidus transitório no pós-operatório P. compatível com craniofaringeoma Iniciado tratamento com GH recombinante, (0,1 U/Kg, 06 x/sem). Após 16 meses apresentou no CT imagem sugestiva de recidiva do tumor Encaminhado para radio- terapia (acelerador linear). Com a idade de 16 anos e 02 meses foi reavaliado quanto à resposta das gonadotrofinas hipofisárias ao estímulo com GnRH, após “priming” com testosterona, mostrando falta de resposta hipofisária. Os demais setores avaliados, à exceção do somatotrófico, apresentaram resposta normal.
. Após 16 meses apresentou no CT imagem sugestiva de recidiva do tumor. Encaminhado para radio- terapia (acelerador linear). Com a idade de 16 anos e 02 meses foi reavaliado quanto à resposta das gonadotrofinas hipofisárias ao estímulo com GnRH, após priming com testosterona, mostrando falta de resposta hipofisária. Os demais setores avaliados, à exceção do somatotrófico, apresentaram resposta normal.")
8
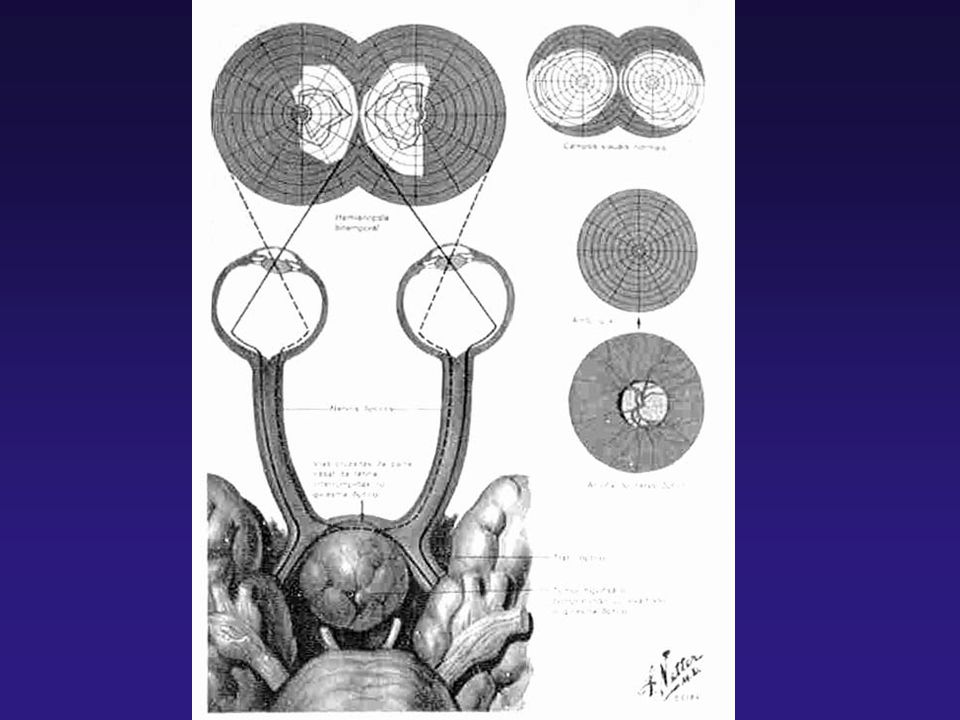
TUMORES HIPOFISÁRIOS CLÍNICA - TUMORES NÃO FUNCIONANTES GERALMENTE SÃO DIAGNOSTICADOS PELA PRESENÇA DE SINAIS / SINTOMAS NEUROLÓGICOS (HIPERTENSÃO INTRA-CRANIANA, CEFALÉIA, ALTERAÇÃO DE CAMPO VISUAL) OU PELA AUSÊNCIA DE PRODUÇÃO HIPOFISÁRIA
 OU PELA AUSÊNCIA DE PRODUÇÃO HIPOFISÁRIA")
11
HIPOFUNÇÃO DE GLÂNDULA ALVO
- PRIMÁRIA: a lesão está na glandula alvo - SECUNDÁRIA: a lesão está na hipófise - TERCIÁRIA: a lesão está no hipotálamo

12
DEFICIÊNCIAS HIPOFISÁRIAS
GH ACTH TSH PROLACTINA LH FSH ADH

13
DEFICIÊNCIA DE GH CAUSAS . INFECÇÕES PRÉ-NATAIS
. DISGENESIA / AGENESIA HIPOFISÁRIA . SELA VAZIA . DOENÇAS INFLAMATÓRIAS DO SNC . TRAUMATISMOS CRANIANOS. . MALFORMAÇÕES SNC . HIPOPITUITARISMO IDIOPÁTICO . TUMORES CRANIANOS

15
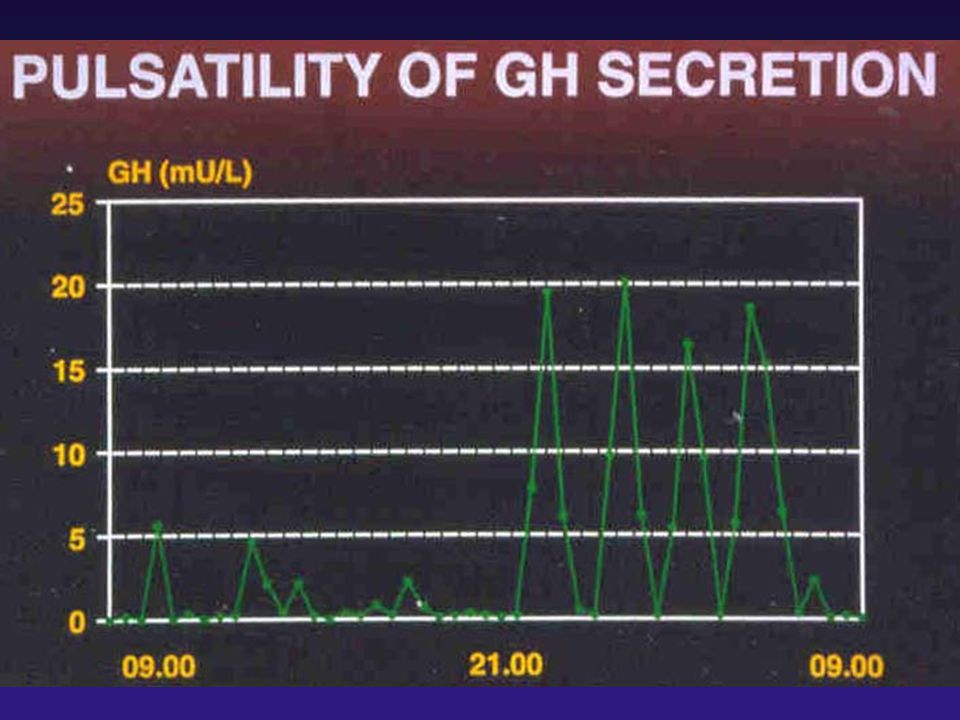
CONTROLE DA SECREÇÃO DE GH
PRINCIPAL ESTÍMULO DA SECREÇÃO DE GH: GHRH - OUTROS ESTÍMULOS: exercício físico, sono fisiológico, hipoglicemia, arginina, clonidina,

16
CONTROLE DA SECREÇÃO DE GH - PRINCIPAL INIBIDOR: SOMATOSTATINA - OUTROS INIBIDORES: HIPERGLICEMIA BROMOCRIPTINA

17
DEFICIÊNCIA DE GH CLÍNICA: . HIPOGLICEMIA (CIANOSE / CONVULSÃO)
. BAIXA ESTATURA PROPORCIONAL . DIMINUIÇÃO DA VELOCIDADE DE CRESCIMENTO . HIPOGLICEMIA (CIANOSE / CONVULSÃO) . MICROPÊNIS / MICRORQUIDIA / CRIPTORQUIDIA . VOZ AGUDA . ACÚMULO DE GORDURA INFRA-UMBILICAL . ASSOCIAÇÃO A SINAIS / SINTOMAS DE + DEFICIÊNCIAS
 . MICROPÊNIS / MICRORQUIDIA / CRIPTORQUIDIA. . VOZ AGUDA. . ACÚMULO DE GORDURA INFRA-UMBILICAL. . ASSOCIAÇÃO A SINAIS / SINTOMAS DE + DEFICIÊNCIAS.")
18
DEFICIÊNCIA DE GH: DIAGNÓSTICO
IGF-1 - IGFBP-3 TESTES PROVOCATIVOS . CLONIDINA . HIPOGLICEMIA INDUZIDA (ITT)
")
19
DEFICIÊNCIA DE GH: TERAPIA
Reposição de GH

20
TUMORES HIPOFISÁRIOS - MICROADENOMAS - MACROADENOMAS

21
TUMORES HIPOFISÁRIOS CLÍNICA -TUMORES SECRETANTES SÃO DIAGNOSTICADOS
PELO QUADRO CLÍNICO DE EXCESSO DE PRODUÇÃO DE UM HORMÔNIO

22
TUMORES HIPOFISÁRIOS PRODUTORES DE PROLACTINA PRODUTORES DE ACTH
PRODUTORES DE GH PRODUTORES DE TSH NÃO FUNCIONANTES

23
HIPERPROLACTINEMIA Prof. Dr. Heraldo Mendes Garmes FCM-UNICAMP

24
HIPERPROLACTINEMIA PREVALÊNCIA
0,4% na população geral adulta 50% dos tumores hipofisários secretores

25
SECREÇÃO DE PROLACTINA
serotonina dopamina PRL + (-) TRH hipófise mamas
 TRH. hipófise. mamas.")
26
CAUSAS DE HIPERPROLACTINEMIA
Fisiológicas Farmacológicas Patológicas Idiopática

27
CAUSAS PATOLÓGICAS DE HIPERPROLACTINEMIA
Desordens hipotalâmicas ou da haste hipofisária Tumores hipofisários Hipotiroidismo Irritação da parede torácica

28
QUADRO CLÍNICO Sintomas da disfunção hormonal: hipogonadismo, diminuição da libido, galactorréia,impotência, infertilidade, redução da massa óssea Sintomas do efeito de massa no SNC: cefaléia, alteração do campo visual, epilepsia, hidrocefalia, oftalmoplegia

29
DIAGNÓSTICO-IMAGEM lesão grande - nível de prolactina maior

30
TRATAMENTO Objetivos: diminuir massa tumoral preservar hipófise normal
restaurar conseqüências clínicas Drogas, cirurgia e radioterapia

31
TRATAMENTO- CLÍNICO Agonista dopaminérgico: Bromocriptina Lisuride
Cabergolina Análago da somatostatina

32
TRATAMENTO - CIRURGIA Cura hormonal a longo prazo: micro: 64 a 74%
macro: 7 a 26% Indicações: intolerância ou resistência à droga crescimento tumoral

33
RADIOTERAPIA Tipos: externa: desaconselhável radiocirurgia: ?
Indicação para radiocirurgia: remoção cirúrgica incompleta que não responde ao tratamento clínico

34
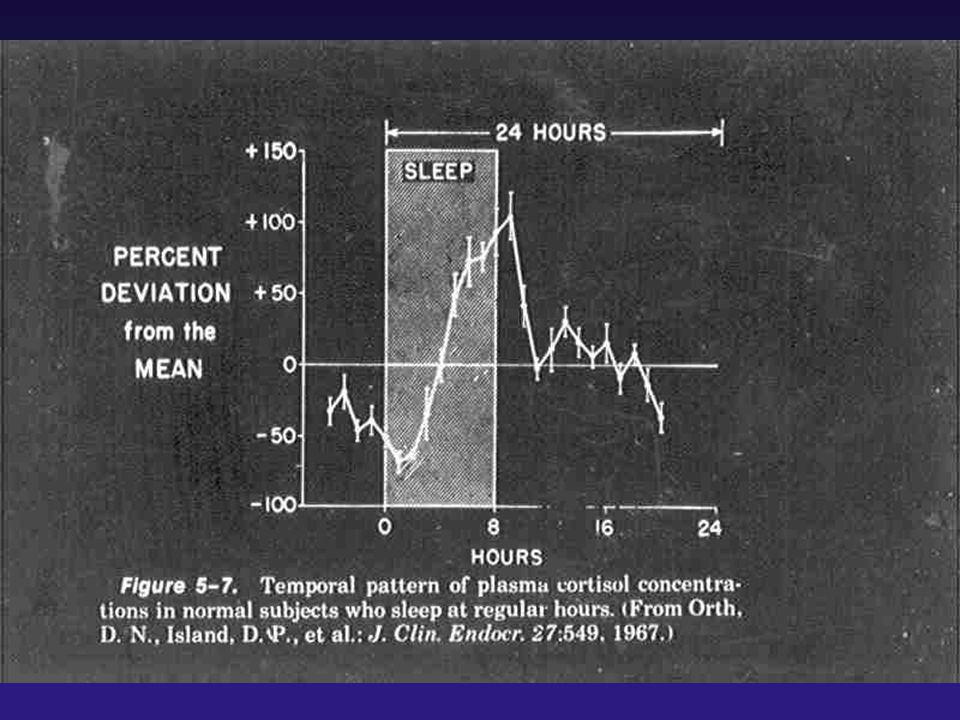
SÍNDROME DE CUSHING

35
CONTROLE DA SECREÇÃO DE ACTH - ESTIMULAM A SECREÇÃO CRH, AVP, Adrenalina, Serotonina, Acetilcolina, Ocitocina Angiotensina, - INIBEM A SECREÇÃO Glicocorticóides, GABA, Beta-endorfina, Dopamina

37
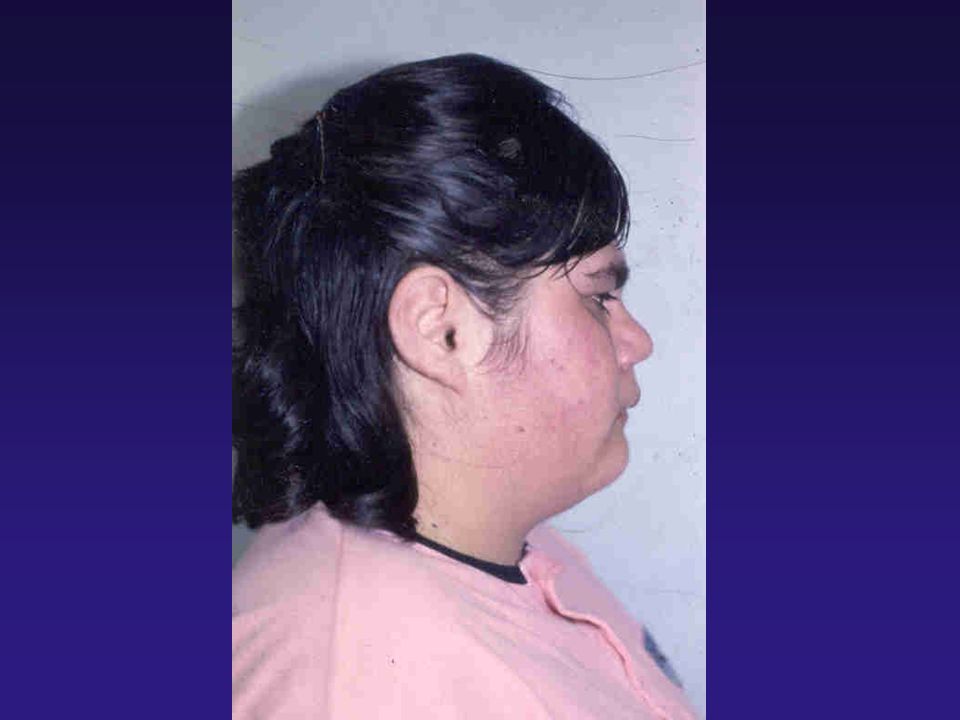
TUMORES HIPOFISÁRIOS PRODUTORES DE ACTH
CLÍNICA: - AUMENTO DE PESO / OBESIDADE CENTRAL - HIPERTENSÃO - HIRSUTISMO - ESTRIAS VIOLÁCEAS - HIPERPIGMENTAÇÃO - ACNE - INTOLERÂNCIA À GLICOSE / DIABETES

40
SUPRESSÃO ACTH COM DEXAMETASONA
- DOSE ÚNICA: cortisol plasmático basal e pós 1 mg às 24 h. - DOSE BAIXA: ACTH e cortisol plasmático basais e após 2 mg/d/2d e cortisol urinário de 24 h. basal e durante os 2 dias de dexa - DOSE ALTA: ACTH e cortisol plasmático basais e após 8 mg/d/2d e cortisol urinário de 24 h. basal e durante os 2

41
SÍNDROME DE CUSHING TERAPIA
LOCALIZAÇÃO DO TUMOR E CIRURGIA ESPECÍFICA: Adrenal Hipofisária Ectópica

42
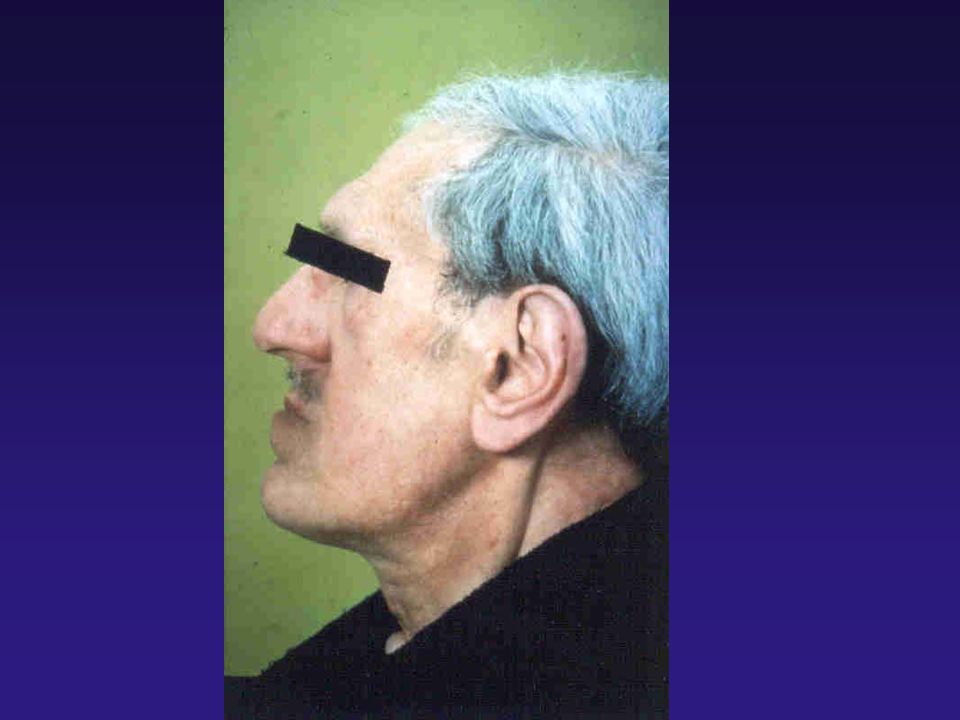
Campanha de incentivo ao diagnóstico de Acromegalia - Akhenaton

43
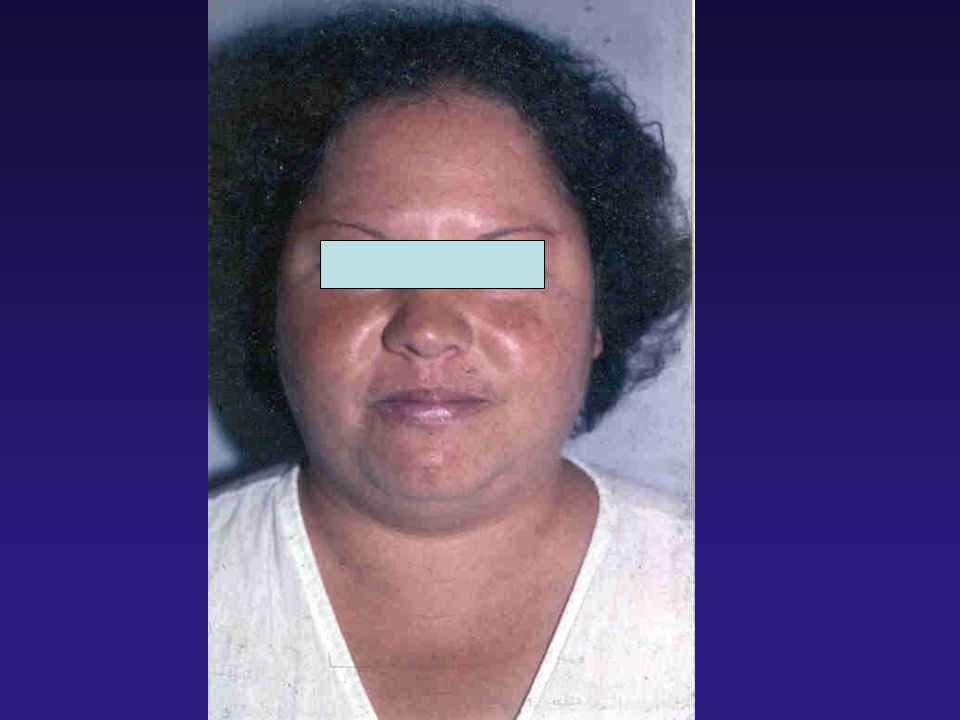
CASO CLÍNICO I: MG, 34 anos, branca, feminina, natural de Belo Horizonte, procedente de São carlos. Q.P.: Dores nas articulações dos joelhos, de caráter não inflamatório, há 2 anos. I.C.: Aumento das extremidades, com dificuldade para colocar anéis, e aumento do número dos calçados (de 36 para 39) nos últimos anos. Episódios de cefaléia holocraniana, diária a 1 ano. E.F.: BEG, corada, hidratada, afebril e sem edema. Nítido aumento das extremidades, com espessamento de coxim calcâneo Fácieis grosseiro, prognatismo e afastamento dos dentes, em arcada inferior. PA = 160/90 mmHg; Semiologia cárdio pulmonar normal. Abdomen flácido, fígado palpável a 2 cm da BCD.
 nos últimos anos. Episódios de cefaléia holocraniana, diária a 1 ano. E.F.: BEG, corada, hidratada, afebril e sem edema. Nítido aumento das extremidades, com espessamento de coxim calcâneo Fácieis grosseiro, prognatismo e afastamento dos dentes, em arcada inferior. PA = 160/90 mmHg; Semiologia cárdio pulmonar normal. Abdomen flácido, fígado palpável a 2 cm da BCD.")
44
Exames laboratoriais: hemograma normal ; glicemia = 154 mg/dl;
GTT COM DOSAGEM DE GH: Tomografia computadorizada de sela túrcica: Aumento de volume selar com lesão de 2,5x2,0x3,0 cm, compatível com macroadenoma hipofisário. tempo HGH (ng/ml) Glicemia (mg/dl) 45 148 30 58 174 60 63 210 90 61 230 120 198 180 50 178
 Glicemia (mg/dl)")
45
Evolução: Hipofisectomia com retirada de tumor hipofisario Imunohistoquímica: tumor produtor de GH. Níveis de GH ao teste de estímulo com TRH + GnRH Indicada radioterapia de região selar A paciente iniciou uso de L-tiroxina (100mcg/dia), prednisona (7,5 mg/dia) e reposição estrogênica. tempo HGH (ng/ml) TSH (mU/ml) LH (mU/ml) FSH (mU/ml) 12 0,2 1,5 2,2 15 18 0.4 2,0 3,0 30 25 0,5 2,5 45 20 1,8 60 22 0,3 1,9
, prednisona (7,5 mg/dia) e reposição estrogênica. tempo. HGH (ng/ml) TSH (mU/ml) LH (mU/ml) FSH (mU/ml) 12. 0,2. 1,5. 2, ,0. 3, ,5. 2, , ,3. 1,9.")
46
Acromegalia Prevalência: 50 a 70 casos por milhão de pessoas
Doença insidiosa de difícil diagnóstico Prevalência: 50 a 70 casos por milhão de pessoas Incidência: 3,3 casos novos por milhão de pessoas a cada ano Idade ao diagnóstico: anos em média

47
Acromegalia Fatores associados ao aumento da mortalidade: Mortalidade
Aumento de 2 a 4 vezes comparado com a população geral Fatores associados ao aumento da mortalidade: Aumento dos níveis de GH e suas complicações Dificuldade de diagnóstico (doença insidiosa) Idade avançada ao diagnóstico Doenças associadas (cardiopatias, diabetes, HAS)
 Idade avançada ao diagnóstico. Doenças associadas (cardiopatias, diabetes, HAS)")
49
Por que um acromegálico procura um clínico?
Dores articulares Cefaléia Sintomas de diabetes Hipertensão arterial Diminuição do campo visual

50
Condições clínicas associadas à Acromegalia
Edema de tecidos moles, espaçamento dentário Skin tags, sudorese Cardiopatia, hipertensão Apnéia do sono Neoplasias, pólipos intestinais Diabetes Sínd. do túnel do carpo Artralgia e artrite Alterações sexuais Aumento do número dos calçados Thorner MO et al, 1992

51
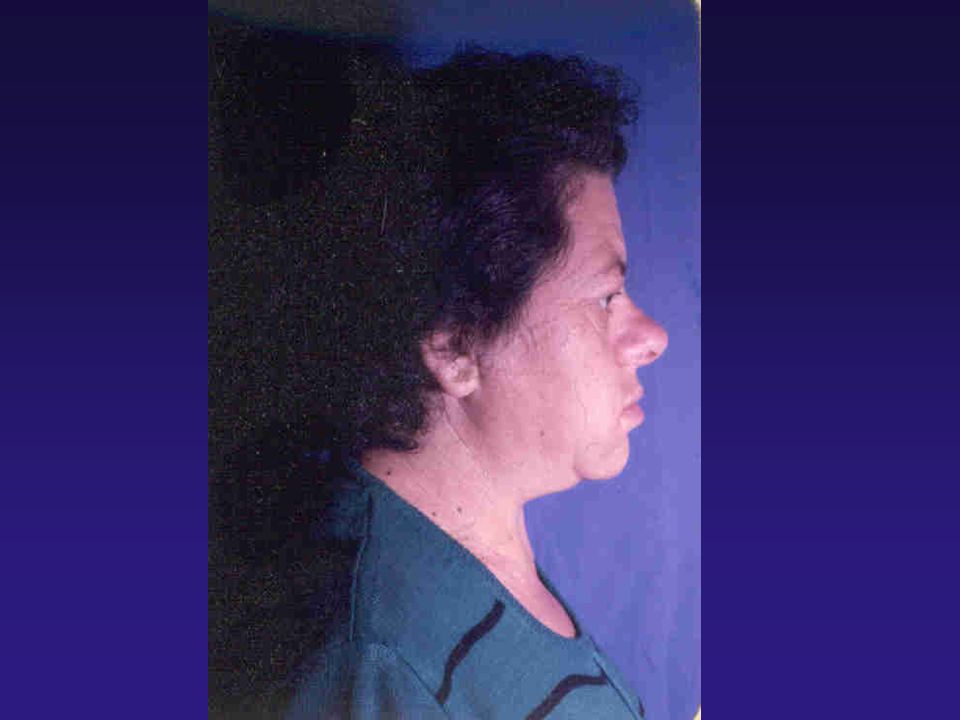
Paciente na juventude

52
Anos após o desenvolvimento da Acromegalia

53
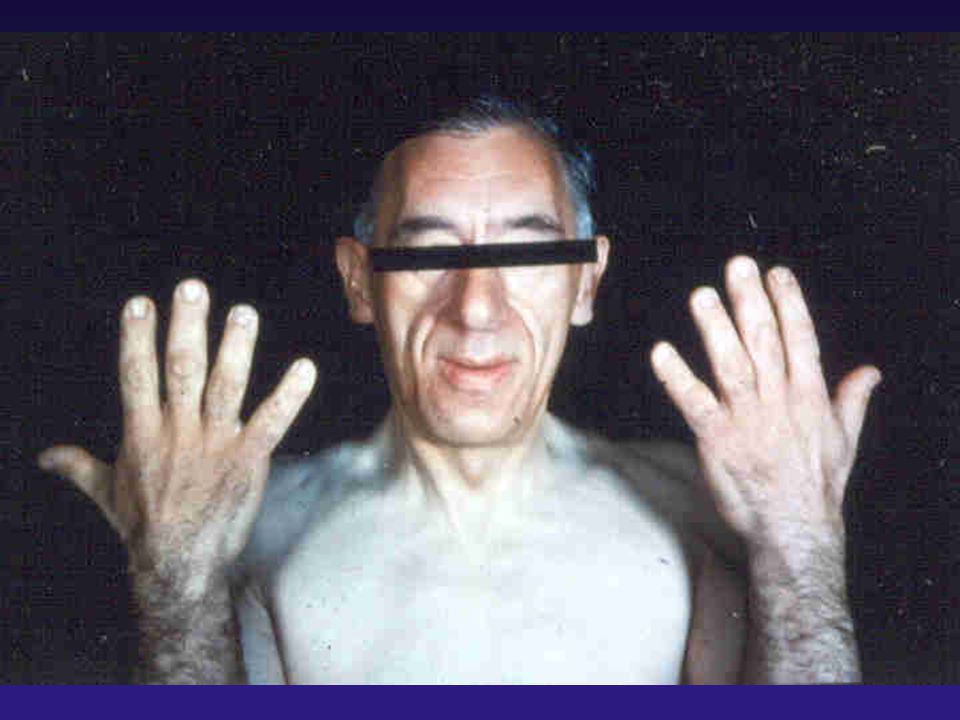
Mãos de um acromegálico

58
Artropatia (geralmente a artropatia é não inflamatória)
Na primeira consulta 60 a 70 % dos pacientes apresentam envolvimento de grandes articulações periféricas (ombro, joelho, quadril) e 50 % apresentam artropatia axial (coluna lombar). (geralmente a artropatia é não inflamatória) Causa: Nível elevado de GH e principalmente de IGF-I Barkan A., Pituitary, 2001
 e 50 % apresentam artropatia axial (coluna lombar). (geralmente a artropatia é não inflamatória) Causa: Nível elevado de GH e principalmente de IGF-I. Barkan A., Pituitary,")
59
Acometimento de extremidades

60
Acometimento de extremidades

61
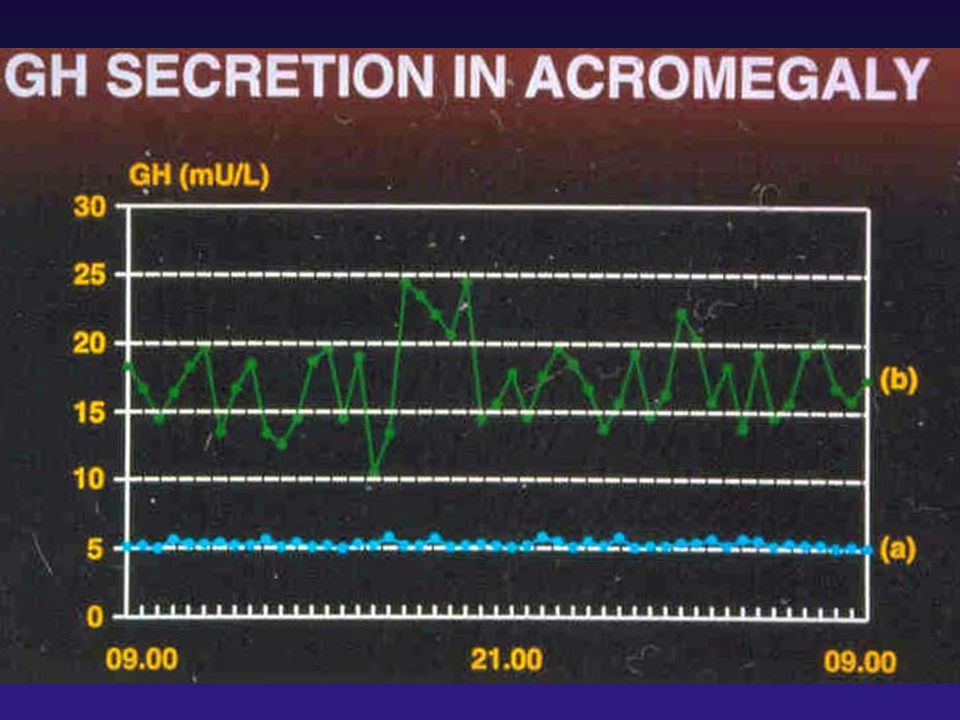
TESTE DE SUPRESSÃO DE GH
GTT ORAL - RESPOSTA NORMAL: TESTE DE SUPRESSÃO POSITIVO - TESTE DE SUPRESSÃO NEGATIVO

62
Diagnóstico de acromegalia
Suspeita clínica Avaliação laboratorial inicial GH e IGF-I séricos GH < 0,4 ng/mL e IGF-I normal para idade e sexo GH > 0,4 ng/mL e/ou IGF-I alto Avaliação para confirmação: TTGO GH < 1 ng/mL em qualquer tempo GH > 1 ng/mL em todos os tempos Exclui acromegalia Confirma acromegalia Giustina et al Consensus JCEM

63
Tratamento Cirurgia Radioterapia Tratamento farmacológico

64
Tratamento cirúrgico Taxa de cura: Microadenomas (<1cm): 80-90 %
Macroadenomas (>1cm): < 50 % Giustina et al Consensus JCEM
: < 50 % Giustina et al 2000 Consensus JCEM")
65
Radioterapia Resultado: observado 10 anos após o tratamento
Principal complicação: hipopituitarismo em cerca de 50% dos casos Giustina et al Consensus JCEM

66
Tratamento clínico Análogos da somatostatina:
Octreotida (Sandostatin® LAR®) Lanreotide (Somatuline® SR e Autogel)
 Lanreotide (Somatuline® SR e Autogel)")
67
O que fazer diante de um paciente com suspeita de Acromegalia?
Solicitar a dosagem de: GH (hormônio do crescimento) IGF-I (somatomedina C)
 IGF-I (somatomedina C)")
68
ENCAMINHE O PACIENTE PARA UM ENDOCRINOLOGISTA
Se: GH > 0,4 ng/mL e/ ou IGF-1 estiver acima do valor normal para idade e sexo: ENCAMINHE O PACIENTE PARA UM ENDOCRINOLOGISTA (Caso contrário, o diagnóstico pode ser descartado) Caso estas dosagens hormonais não sejam disponíveis em seu serviço, encaminhe o paciente diretamente para um endocrinologista.
 Caso estas dosagens hormonais não sejam disponíveis em seu serviço, encaminhe o paciente diretamente para um endocrinologista.")
Apresentações semelhantes