Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Grupo de Neurologia Cognitiva e do Comportamento
III SIMPÓSIO INTERNACIONAL DE VIGILÂNCIA DAS DOENÇAS DE TRANSMISSÃO HÍDRICA E ALIMENTAR 21 de Novembro de 2005 Centro de Convenções Rebouças, São Paulo, SP, Brasil. Ricardo Nitrini Grupo de Neurologia Cognitiva e do Comportamento Departamento de Neurologia da FMUSP

2
Nova Variante da Doença de Creutzfeldt-Jakob

3
Doenças Priônicas: Encefalopatias Espongiformes Transmissíveis
Grupo de doenças caracterizadas por progressiva vacuolização do córtex cerebral, morte neuronal e proliferação glial Presença no SNC de uma proteína anormal resistente a proteases denominada prion

4
DOENÇAS PRIÔNICAS Acometem animais e seres humanos Transmissíveis
Material transmissor não é inativado por processos que inativam ácidos nucleicos Podem ser hereditárias (e, simultaneamente, transmissíveis)
")
5
DOENÇAS PRIÔNICAS HUMANAS
Kuru Doença de Creutzfeldt-Jakob Doença de Gerstmann-Sträussler-Scheinker Insônia Fatal Familial Variante da Doença de Creutzfeldt-Jakob

9
D. Carleton Gajdusek (1923- )
Prêmio Nobel 1976

10
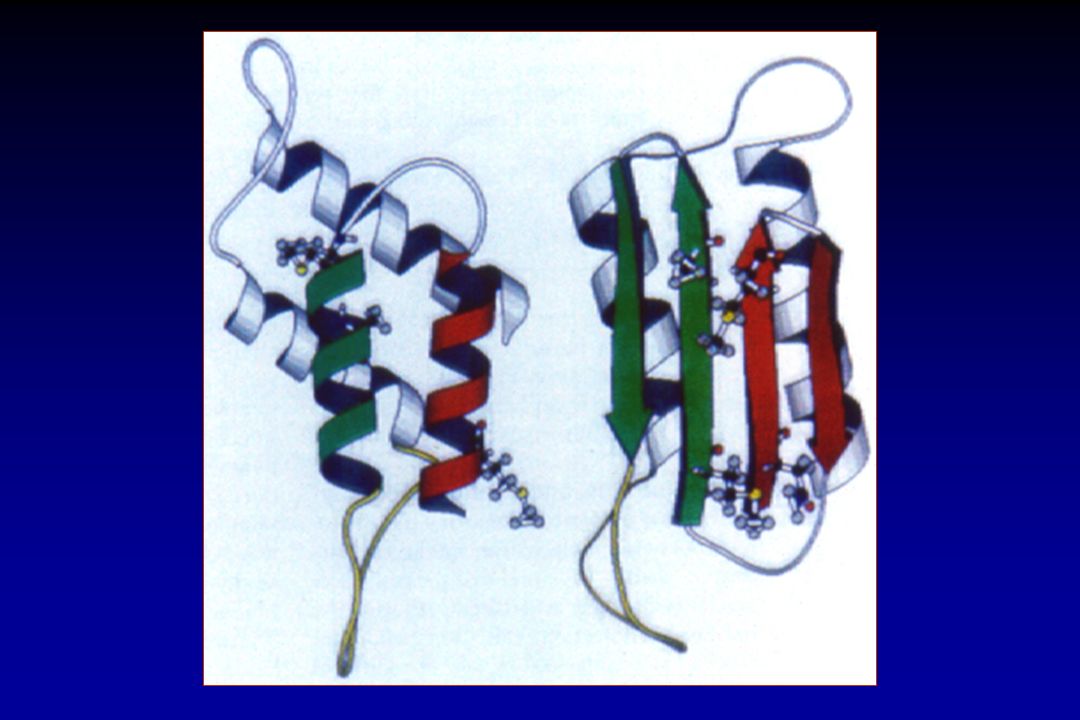
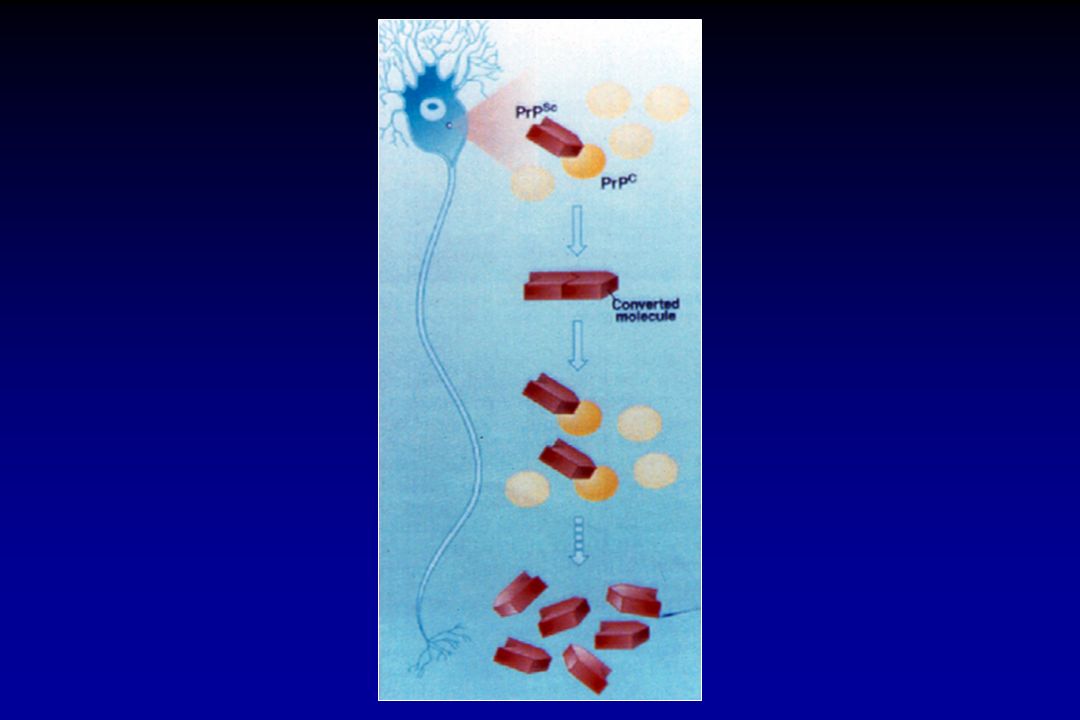
DOENÇAS PRIÔNICAS A principal hipótese patogênica baseia-se na transfor- mação de uma proteína normal (PrPC) em uma isoforma estruturalmente anormal (PrPSc), parcialmente resistente a proteases. A PrPSc atua como uma fôrma (template), transformando mais unidades de PrPC em PrPSc, que se acumulam no interior da célula, destruindo-a.
 em uma isoforma estruturalmente anormal (PrPSc), parcialmente resistente a proteases. A PrPSc atua como uma fôrma (template), transformando mais unidades de PrPC em PrPSc, que se acumulam no interior da célula, destruindo-a.")
13
Stanley B. Prusiner (1942- ) Prêmio Nobel 1997
 Prêmio Nobel 1997")
14
Predominância de homozigose
Polimorfismo no codon 129 Pop. Caucasiana: 38% M/M 51% M/V 11% V/V DCJ esporádico DCJ iatrogênico Predominância de homozigose Está relacionado com a variabilidade do quadro clínico, da evolução e dos achados anatomopatológicos.

15
Classificação das Doenças Priônicas Humanas
Esporádicas Hereditárias Adquiridas

16
Doenças Priônicas Humanas Esporádicas
Doença de Creutzfeldt-Jakob (Insônia Fatal - Forma esporádica)
")
17
Doença de Creutzfeldt-Jakob Esporádica
85 -90% casos Igual prevalência entre os sexos Incidência 1 : hab./ ano Idade média de início: 60 anos Duração média: 8 meses

18
Anatomia Patológica: DCJ

19
Anatomia Patológica: DCJ

20
Imunohistoquímica com anticorpos anti-PrP (3F4)
DCJ: Imunohistoquímica com anticorpos anti-PrP (3F4)
")
21
EEG típico de DCJ: Atividade periódica curta

24
Doenças Priônicas Humanas Hereditárias
D. de Creutzfeldt-Jakob familial Doença de Gerstmann-Sträussler-Scheinker Insônia Fatal Familial Atípicas

25
Doença de Creutzfeldt-Jakob Iatrogênica
Transplante de córnea Hormônio de crescimento de cadáveres Aloenxertos de dura-máter Eletrodos intracorticais

26
NOVA VARIANTE DA DOENÇA DE CREUTZFELDT - JAKOB

27
Nova Variante da DCJ primeiros casos de Encefalopatia espongiforme bovina (BSE) proibido uso de ração com proteína animal; notificação compulsória de BSE

28
Nova Variante da DCJ Centro de controle de unificado para doenças priônicas humanas na GB Casos de DCJ atípicos (nvDCJ) Demonstrado vínculo nvDCJ-BSE

29
Will, R. World Congress of Neurology, Sydney, November 2005.

30
vDCJna Grã-Bretanha (até outubro de 2005) N=152
2005 (4 de novembro) 4
 4.")
31
vDCJ fora da Grã-Bretanha (até outubro de 2005)
Outros 10 países: França (9) USA Portugal Itália Holanda Irlanda Espanha Japão Canadá Arábia Saudita
 USA. Portugal Itália. Holanda Irlanda. Espanha Japão. Canadá Arábia Saudita.")
32
Diferenças entre a forma esporádica e a nova variante da DCJ
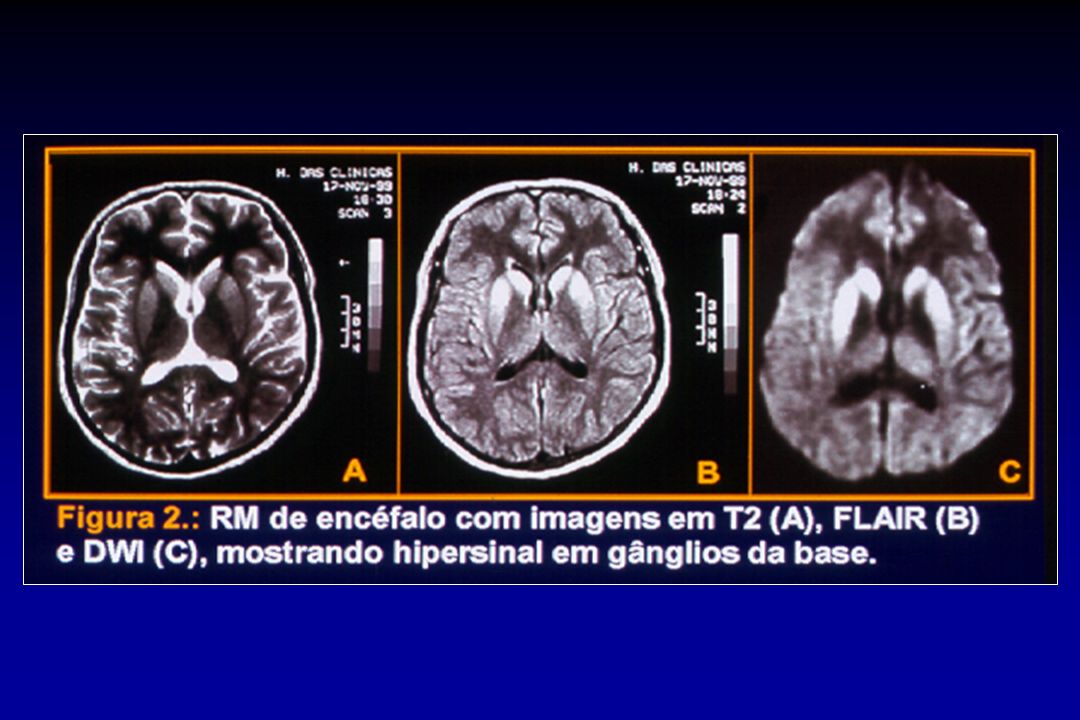
Média de idade de óbito 66 anos 29 anos Mediana da duração 4 meses 13 meses Hipersinal na RM Caudado e putâmen (60%) Tálamo (pulvinar) (90%) no LCR Mais de 90% 50% Imunohistoquímica nas tonsilas Negativa Positiva EEG Típico (70%) Típico (0%) Will, R. World Congress of Neurology, 2005
 Tálamo (pulvinar) (90%) no LCR. Mais de 90% 50% Imunohistoquímica nas tonsilas. Negativa. Positiva. EEG. Típico (70%) Típico (0%) Will, R. World Congress of Neurology,")
33
Collie et al. AJNR, 2003

34
Diferenças entre a forma esporádica e a nova variante da DCJ
Sintomas psiquiátricos precoces Incomum Comum Sintomas sensitivos dolorosos Ataxia cerebelar tardia Todos Demência Precoce Tardia Polimorfismo no codon 129 Homo e heterozigose 100% MM Patologia Placas (10%) Placas floridas (100%) Will, R. World Congress of Neurology, 2005;Johnson, RT. Lancet Neurology 2005.
 Placas floridas (100%) Will, R. World Congress of Neurology, 2005;Johnson, RT. Lancet Neurology")
35
Placa “Florida” na nova variante DCJ

36
Critérios Diagnósticos para a vDCJ
I A. Transtorno neuropsiquiátrico progressivo B. Duração da doença > 6 meses C. Investigação não sugere outra doença D. Sem história de exposição iatrogênica E. Sem história familial de d. priônica II A. Sintomas psiquiátricos precoces B. Sintomas dolorosos persistentes C. Ataxia D. Mioclonia ou coréia ou distonia E. Demência III. A. EEG atípico ou não realizado B. Hipersinal no pulvinar na RM IV. A. Biópsia de tonsila positiva para PrPSc DEFINITIVO I e comprovação neuropatológica de vDCJ PROVÁVEL I e 4/5 de II e III A e III B OU I e IV A POSSÍVEL I e 4/5 de II e III A Will, R. World Congress of Neurology, 2005

37
Hipótese diagnóstica inicial em 150 casos comprovados de vDCJ
% Depressão 76 50,7 “Caso neurológico” 27 18,0 Diagnóstico incerto 19 12,7 Outros* 12 8,0 Ansiedade 7 4,7 “Stress” 4 2,7 Transtorno psicótico 2 1,3 Transtorno funcional 1 0,7 Sem informação TOTAL 150 100,0 * Outros: problema ortopédico (3), febre glandular (3), asma, anorexia nervosa, reação de luto, problema odontológico, problema endócrino, doença viral
, febre glandular (3), asma, anorexia nervosa, reação de luto, problema odontológico, problema endócrino, doença viral.")
38
Causa mortis (neuropatologicamente descartados) DCJ esporádica
Freqüência N=40) DCJ esporádica 7 Doença de Alzheimer Sem diagnóstico definitivo 5 D. paraneoplásica/neoplasia maligna 3 Encefalite viral Demência frontotemporal 2 Doença desmielinizante Panencefalite esclerosante subaguda Fibrose miocárdica Doença cerebrovascular 1 Transtorno metabólico Atrofia de mútiplos sistemas Doença de Huntington Doença de Wilson Encefalite límbica autoimune Encefalite de origem indeterminada
 DCJ esporádica. 7. Doença de Alzheimer. Sem diagnóstico definitivo. 5. D. paraneoplásica/neoplasia maligna. 3. Encefalite viral. Demência frontotemporal. 2. Doença desmielinizante. Panencefalite esclerosante subaguda. Fibrose miocárdica. Doença cerebrovascular. 1. Transtorno metabólico. Atrofia de mútiplos sistemas. Doença de Huntington. Doença de Wilson. Encefalite límbica autoimune. Encefalite de origem indeterminada.")
39
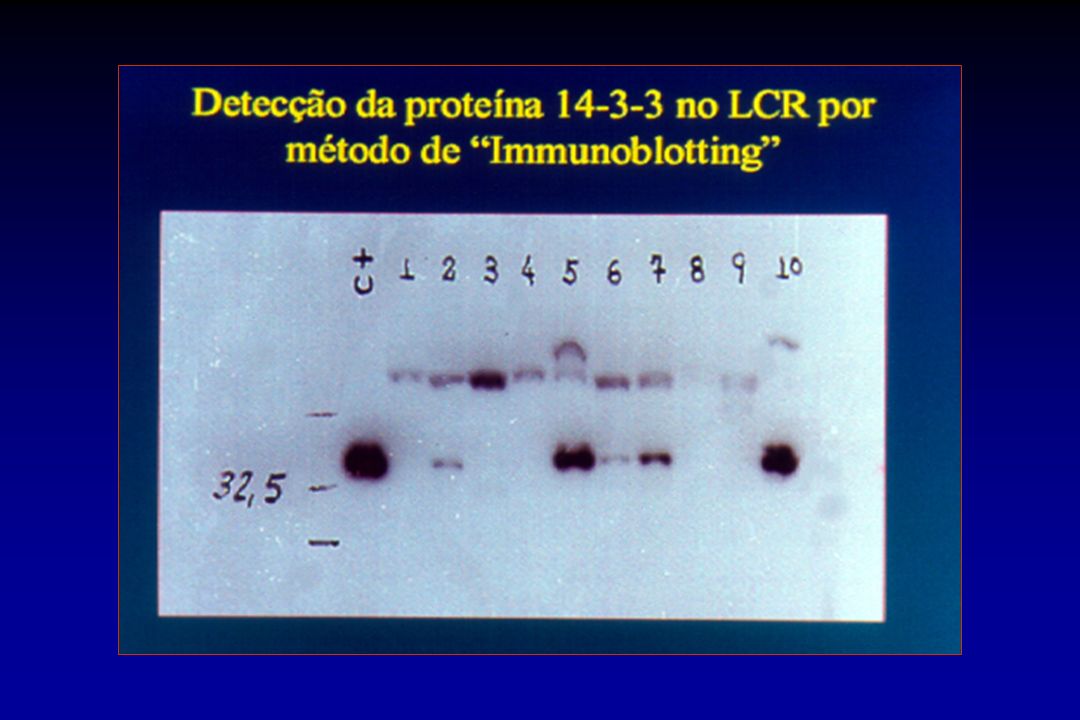
Exames complementares na vDCJ
RM: Hipersinal no pulvinar com técnica de difusão – positiva em 97/106 casos comprovados (92%) Biópsia de tonsila: positiva em 18/18
 Biópsia de tonsila: positiva em 18/18.")
40
Transmissão da vDCJ Não há evidência de transmissão placentária: 9 casos de parto na vigência de vDCJ. Crianças sem a doença, com idades < 10 anos (Will, 2005) Um caso de vDCJ pós-transfusão (Llewelyn et al., Lancet 2004) Outro caso pós-transfusão com PrPSc no baço e linfonodo cervical (sem doença clínica; era heterozigoto no codon 129)
 Um caso de vDCJ pós-transfusão (Llewelyn et al., Lancet 2004) Outro caso pós-transfusão com PrPSc no baço e linfonodo cervical (sem doença clínica; era heterozigoto no codon 129)")
41
TRATAMENTO Não há tratamento recomendado para as doenças priônicas. Para as adquiridas, prevenção é essencial Tem sido proposto o emprego de quinacrina e flupirtina para a DCJ mas os efeitos são muito discretos e/ou duvidosos

42
TRATAMENTO Maleato de Flupirtine Quinacrina + Clorpromazina na DCJ
Analgésico não-opióide Efeito citoprotetor in vitro e in vivo sobre neurônios que foram induzidos à apoptose. Quinacrina + Clorpromazina na DCJ

44
E o H5N1V? Coma mais peixe?

45
FIM

Apresentações semelhantes










 NO ESTADO DE SÃO PAULO NA DÉCADA DE 90.>")




 NO ESTADO DE SÃO PAULO NA DÉCADA DE 90.>")






