Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Como o genoma humano foi sequenciado
Elisa Mota Filipe Dutra Frederico Kremer Rafael Woloski Vinícius da Rosa

2
Por que seqüenciar o genoma humano?
Determinar as possíveis causas para doenças genéticas Desenvolver terapias gênicas Obesidade Hipertensão Diabetes

3
Histórico 1984: Cientistas se reúnem em Utah para debater sobre as maneiras de identificar mutações dos sobreviventes às bombas atômicas de Hiroshima e Nagasaki

4
1987: Relatórios do Departamento de Energia americano recomendavam que o governo realizasse um esforço para determinar a seqüência do genoma humano

5
1990: Lançamento do Projeto Genoma Humano, tendo Estados Unidos, Japão, França, Alemanha, China e Reino Unido como os centros do projeto.

6
1994: Mapa genômico humano detalhado

7
1996: Reunião de cientistas do mundo todo para discutir métodos de seqüenciamento e discutir os rumos da liberação dos dados; Começa o seqüenciamento de fato do genoma humano.

8
1999: Celera entra na corrida no seqüenciamento do genoma humano

9
2001: O primeiro rascunho do genoma humano foi lançado ao mesmo tempo pela Celera e pelo Projeto Genoma Humano

10
2003: Projeto Genoma Humano concluído
2004:

11
Projeto Genoma Humano Intuito de mapear o genoma humano, identificando genes responsáveis por características “normais” e patológicas. Assim, levar avanços à área médica e prevenir características indesejáveis, como doenças. Vacinas de DNA poderiam ser desenvolvidas e remédios seriam adequados ao “perfil genético” de indivíduos. Modelos animais mais usados também foram analisados, para pesquisas posteriores.

12
Análises Método de Sanger; Seqüenciamento hierárquico por shotgun
Eletroforese em gel acrilamida; Leitura da seqüência;

13
Divulgação (2001)
")
14
Abril de 2003: Conclusão.

15
Iniciativa privada:

16
Celera Genomics é um empresa do setor farmacêutico-biotecnológico que desenvolve tecnologias de diagnóstico e tratamento. Fundada em 1998, a empresa tinha, dentre seus objetivos, o sequenciamento de genoma humano para fins comerciais. Foi idealizada por Craig Venter e pela Applera corporation.

17
Antes do Sequenciamento do Genoma Humano a Celera já havia ajudado no sequenciamento do genoma da Drosophila Megalogaster. O projeto foi realizado em parceria com o consórcio Projeto Genoma Drosophila e serviu de base para os protocolos e métodos utilizados no sequenciamento do Genoma Humano.

18
Origem do material 2 homens e 3 mulheres cederam material para o sequenciamento. Deste indivíduos, um era afro-americano, um era hispânica, um era de origem chinesa e dois era caucasianos. Os indivíduos assinaram termos de consentimento e duas identidades foram ocultadas. Foram retirados 130 mL de sangue de cada um.

19
Construção de bibliotecas de DNA e sequenciamento

20
O processo de extração e purificação visava apenas o DNA cromossômico.
Para a construção da biblioteca, o DNA humano foi clivado em pedaços relativamente pequenos e de forma aleatória, inserido em plamídeos e estes foram inserido em E. Coli. Os plasmídeos apresentavam, no final, tamanhos de 2, 10 ou 50 kb.

21
O DNA das colônias transformadas foi extraído e foi feita a reação de sequenciamento com os ddNTPs.
Para o sequenciamento, foram utilizados seqüenciadores ABI PRISM 3700. Cada amostra seqüenciada era identifica com um código de barras. Desde o começo do projeto, até o fim da etapa de sequenciamento, nem um dia se quer foi desperdiçado. Um alto controle de qualidade durante todo o processo garantiu uma taxa de apenas 1 em cada leituras com qualidade inferior a 98%.

22
Montagem do genoma

23
Whole Genome Shotgun Assembly Compartmentalized Shotgun Assembly
Após o sequenciamento, métodos computacionais foram necessários para a montagem do genoma. Como o genoma havia sido fragmentado em pedaços relativamente pequenos e de forma aleatória, a montagem se torna um processo complexo, sobretudo em zonas de DNA repetitivo. A Celera utilizou dois métodos para o sequenciamento: Whole Genome Shotgun Assembly Compartmentalized Shotgun Assembly

24
Whole Genome Shotgun Assembly

25
Para a montagem do genoma inteiro, foram utilizados programas que já haviam sido utilizados no Projeto Genoma Drosophila. Estes programas consistem em um procedimento denominado Whole-genome Assembly (WGA). A montagem do WGA é feita em cinco passos: Screener. Overlapper. Unitigger. Scaffolder. Repeat Resolver.

26
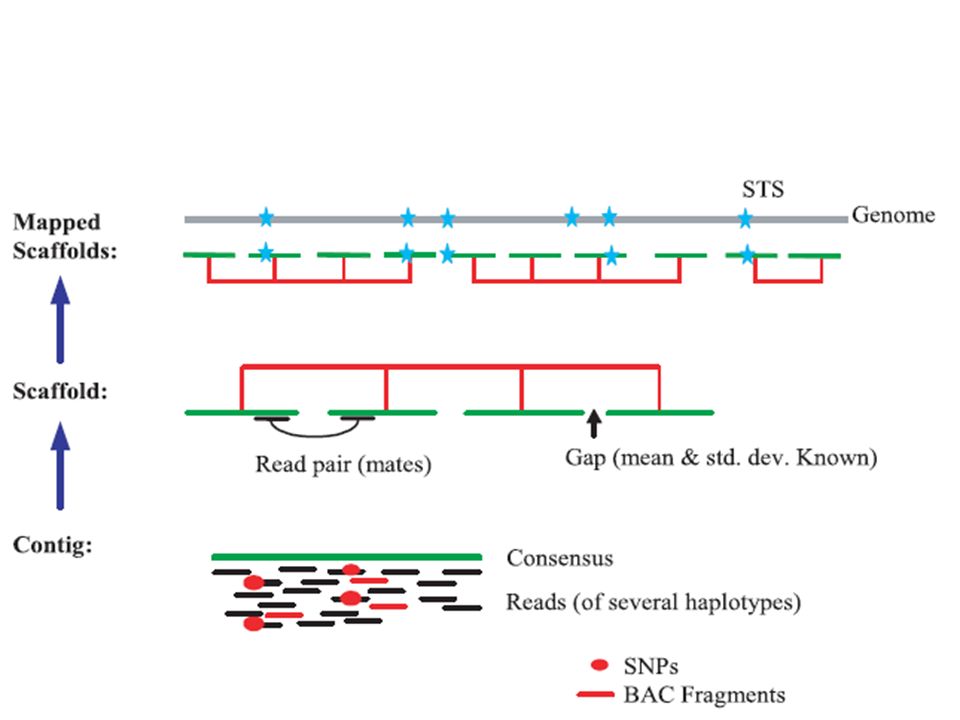
Screener: Este passo consiste em procurar e marcar regiões de microssatélites com elementos de 6 pares de base ou menos. O algoritmo também rastreia zonas de DNA ribossomal e sequencias do tipo Alu e Line. As informações obtidas neste passo serão uteis para a construção de contigs e scaffolds. Contigs: são formados por fragmentos menores que são sobrepostos com a finalidade de se construir um seqüência maior de forma ininterrupta. Scaffolds: Conjunto de contigs.

28
Overlapper: Esta etapa consiste na sobreposição dos fragmentos. O computador comparará cada fragmento com todos os outros fragmentos e selecionará apenas as sobreposições que apresentam menos de 6% de diferença no match. Estatisticamente falando, o processo de sobreposição possui um baixa taxa de erro. Alinhamento errados feitos por coincidência nas sequencias ocorrem em 1 a cada sobreposições. Entretanto, o processo é altamente difícil tendo em vista o grande numero de sequencias a serem comparadas. 40 computadores em paralelo e cada com 4 Gb de RAM e quatro núcleos de processadores levaram cinco dias para terminar a sobreposição.

29
Unitigger: Apesar da etapa de overlapper conseguir criar sequencias consensos de uma grande porção do genoma, ainda existem fragmentos que não conseguem ser sobrepostos de forma correta. Esta etapa utiliza relações estatísticas para descriminar quais fragmentos pertencem a determinadas regiões. Até aqui, aproximadamente 73% do genoma está montado.

30
Scaffolder: As sequencias obtidas nos passos anteriores serão utilizadas na construção de um Scaffold que represente a seqüência do genoma.

31
Repeat Resolution: Um conjunto de três estágios onde a seqüência é analisada com a finalidade de se diminuir a possível taxa de erro. Neste ultimo passo, ocorre uma grande demanda por memória dos computadores. Foram necessários 7 dias de processamento, utilizando-se 10 computadores com processadores de 4 núcleos e 4 Gb de RAM e 16 processadores NUMA com 64 Gb de RAM.

32
Compartmentalized Shotgun Assembly

34
Este processo tem por objetivo complementar as análises obtidas pelo WSA, e consiste na divisão do genoma e grande regiões. Um dos objetivos deste processo é conseguir uma montagem mais “realista” de regiões cromossômicas duplicas e serve para complementar a montagem de determinadas regiões.

35
Para realizar este processo, foram usados dados do genoma que foram gerados pelo consórcio público, denominados PFP data. Os dados dos fragmentos obtidos pelo sequenciamento da Celera, em conjunto com PFP data serão divididos em regiões, denominadas components. A determinação dos components é feita a partir de conhecimentos prévios de analise de sequencias já conhecidas. Como os components são montados de forma independente, regiões duplicadas não serão tratadas com ambigüidades, o que diminui a taxa de erro e a demanda por processamento.

36
Comparação do WSA e CSA

37
Ambos contribuem para uma montagem mais precisa do genoma, cada um utilizando um parâmetro e algoritmos distintos. Agora, com duas montagens diferentes, é possível fazer uma comparação, identificar pontos de concordância e inconsistências durante as montagens. Do ponto de vista estatístico, o CSA é mais “eficiente” que o WSA, visto que na montagens compartimentalizada os erros de cobertura se estendiam por zonas na ordem de mega bases, enquanto a montagem do genoma inteiro possui erros do mesmo tipo que se estendiam por áreas na magnitude de giga bases.

38
A maior precisão da montagens compartimentalizada e sua menor exigência de hardware para processamento fez com que a Celera utilizasse este assembly para as analises posteriores.

39
Com o Assembly selecionado, o próximo passo era a transposição dos scaffolds obtidos pelo CSA nas informações já conhecidas dos cromossomos. Para esta etapa, fora utilizadas informações do GeneMap99, usando-se regiões conhecidas como marcadores para o “encaixe” da seqüência.

40
Completamente seqüenciado?

41
Apesar de toda a exigência, o projeto não conseguiu um cobertura de todo a seqüência do genoma humano. É difícil determinar com certeza, mas uma análise das sequencias de cromossomos já conhecidos, marcadores moleculares, marcadores STS e genomas já conhecidos (como o da Drosophila) leva a crer que as seqüências obtidas pela Celera representam aproximadamente 94% do total.
 leva a crer que as seqüências obtidas pela Celera representam aproximadamente 94% do total.")
42
Análise pós-montagem: predição de genes

43
Desafios da predição de genes:
A predição de genes ainda é um processo impreciso e difícil de ser executado dentro de um genoma grande. Os principais problemas são: Ainda é difícil se definir um parâmetro universal que possa ser transformado em um algoritmo de busca eficiente. Novos algoritmos são necessários para que o computador possa diferenciar melhor as zonas codificadoras das não codificadoras.

44
Com os genes foram identificados:
A identificação dos genes é feitas através de diversas técnicas, dentre elas: Comparação do Genoma com o Genoma de outros organismos, neste caso com o da Drosophila megalogaster e do Rato. Predição automatizada. Utilização de Softwares capazes de identificar genes, como as ferramentas Genscan que apresenta alta sensibilidade na definição de estrutura de genes e foi usada como base para a ferramenta Otto.

45
A ferramenta Otto: Tinha por objetivo emular o processo de identificação de genes que ocorre no organismo. Algoritmo de foca em determinadas regiões do genoma e começa uma análise baseada em padrões já conhecidos de estrutura de genes, fazendo uma busca por características que confirmem se aquela sequencia é codificante. O algoritmo também se mostrou eficiente na identificação de genes que já foram determinados e que tinha suas sequencias disponíveis em bancos de dados, como o RefSeq.

46
Resultado de Identificação:
Utilizando a ferramenta Otto e outras análises complementares a Celera conseguiu identificar genes. Também foram encontrados muitos pseudogenes, regiões que apresentam uma estrutura muito parecida com outros genes mas que, aparentemente, não são expressão.

47
Aplicações Futuras Medicina Molecular Genômica Molecular
Avaliação de Riscos Antropologia e Evolução

48
Medicina Molecular Mudança no foco da medicina de tratar os sintomas para tratar a causa da doença; Medicina com possibilidade de fazer diagnósticos de doenças genéticas mais cedo, mais rápido, com maior precisão e menor custo; Surgimento de novos fármacos e tratamentos que podem prevenir ou até mesmo curar doenças que atualmente são consideradas incuráveis;

49
Modificação ou remoção de seções do genoma que apresentam defeitos em indivíduos;
Melhoramento em genes benéficos para o ser humano, como supressores de tumor; Fármacos com melhor sistema de entrega e melhora no sistema de ação.

50
Genômica Microbiana Desenvolvimento de novas formas de combates de microorganismos patogênicos; Compreender os efeitos de microorganismos benéficos no corpo humano, e testar a adaptação de indivíduos a locais com microbiota diferente, assim como a ação de probioticos.

51
Entender a atividade da microbiota nos seus principais pontos de ação.

52
Avaliação de Riscos Exposição à radiação, mesmo que em curto período de tempo; Exposição à agentes químicos mutagênicos e toxinas causadoras de câncer; Reduzir a probabilidade de transmitir mutações genéticas aos filhos.

53
Compreender o motivo genético de porque certos medicamentos/toxinas tem efeitos variados em diversos tipos de pessoas.

54
Antropologia e Evolução
Estudar a evolução através de mutações da microbiota em diferentes gerações; Estudar a migração de diferentes grupos populacionais baseado na herança genética feminina; Estudar mutações do cromossomo Y para acompanhar gerações e migrações masculinas.

55
Comparar breakpoints na evolução de mutações com a idade da população geral e acontecimentos históricos; Maior compreensão da origem biológica da vida.

57
E mais... Capacidade de determinar o genótipo a baixíssimos custos, em um estudo em que indivíduos poderiam ser testados com aproximadamente marcadores genéticos por menos de R$ ,00; Conseguir monitorar o estado de todas as proteínas em uma única célula durante um experimento;

58
Desafios a serem superados
Número de genes, definir localização e função exatas; Regulação gênica; Organização das sequências; Organização da estrutura cromossômica; Função, tipo, quantidade e distribuição de DNA não-codificante;

59
Coordenação da expressão, síntese de proteínas e eventos pós-transcricionais;
Interação das proteínas nas maquinarias celulares; Diferença entre a função predita de um gene e a função determinada em um experimento; Conservação evolutiva entre organismos;

60
Correlação entre SNPs e diferenças da condição de saúde entre diversos indivíduos;
Prever a suscetibilidade a doenças baseado em variações nas seqüências de um gene; Genes envolvidos em características complexas e doenças multigênicas; Fatores Epigenéticos.

61
Projeto Genoma Humano e Ética
Por Mayana Zatz Banco de DNA: Possibilidade de identificar criminosos X Instabilidade “familiar”; (Baseado em artigo de Dawkins, de 1998, e Estudos de Robert Wright, em seu livro “O animal moral: psicologia evolutiva e vida cotidiana”) Informações sobre tendência a desenvolver determinadas doenças e expectativa de vida: poderiam ser divulgadas a planos de saúde e empregadores. Diagnóstico pré-natal e interrupções de gestação;
 Informações sobre tendência a desenvolver determinadas doenças e expectativa de vida: poderiam ser divulgadas a planos de saúde e empregadores. Diagnóstico pré-natal e interrupções de gestação;")
62
Projeto Genoma Humano e Ética
“Aceitação” de padrões de comportamento ligados a genes X discriminação de indivíduos predispostos à dependência química e comportamentos “indesejáveis”; Possibilidade de escolha do sexo de bebês: “Desbalanceamento sexual” X Prevenção de doenças; Aconselhamento genético: prevenção de doenças X questões familiares; Testes moleculares para detectar doenças “tardias”, ainda sem tratamento; Paradoxo do câncer de mama.

63
Artigos

65
Psoríase A psoríase é uma doença de pele inflamatória comum, com uma etiologia base de fatores ambientais e genéticos. Doença autoimune causa real mal definida. Fatores genéticos contribuem para a suscetibilidade à doença. Suspeita-se: Polimorfismos de nucleotídeo único (SNVS) ou Cópia variantes de número (CNVs)
 ou. Cópia variantes de número (CNVs)")
66
Técnicas Usadas Técnicas genéticas usadas : PCR, PCR em tempo real microarray, e restrição do comprimento do fragmento de análise Suspeita: genes Destaque: ALOX12B, EIF5, CTSC, CDC42EP1, HDGF, MX1, ITM2B, SYNCRIP, MYO5A, ALOX12B, UBE2L6, NAPA, TGM1 e SPRR1A

67
Conclusão Variabilidade na expressão de genes:
Em cada indivíduo idade, raça, sexo, genética, tipos de pele e influências ambientais. Acredita-se que a psoríase é um interferon-imunes e mediada por células T

73
Obrigado!

Apresentações semelhantes