Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Técnicas de Busca e Análise de Genes de Interesse pela internet (in silico)
Antonio Figueira Centro de Energia Nuclear na Agricultura Universidade de São Paulo - Piracicaba, SP

2
Dogma Central

3
Estrutura de Gene Eucarioto

4
Estrutura de Gene Eucarioto

6
Banco de Dados NCBI - National Center for Biotechnology Information
Iniciado em 1988 Missão: melhor entendimento dos processos moleculares que afetam a saúde humana NCBI cria banco de dados públicos e recursos de biologia computacional; e disseminação de informações

8
Histórico de Seqüências do GenBank

9
X Ano Pares Base Sequences
,037,734, X

10
Tipos de Banco de Dados Abrangentes Nucleotídeos Proteínas
GenBank (more...) EMBL: European Molecular Biology Laboratory DDBJ: DNA Data Bank of Japan Proteínas Swiss-Prot Protein Information resource UniProt Estrutura de Proteínas PDB: Protein Data Bank MMDB: Molecular Modeling Database (baseado em PDB) Mapas e Genoma Entrez Genomes
 EMBL: European Molecular Biology Laboratory. DDBJ: DNA Data Bank of Japan. Proteínas. Swiss-Prot. Protein Information resource UniProt. Estrutura de Proteínas. PDB: Protein Data Bank. MMDB: Molecular Modeling Database (baseado em PDB) Mapas e Genoma. Entrez Genomes.")
11
Tipos de Banco de Dados Especializados Organismo-Específico Funcional
Human Genome Sequencing GDB: Genome Database (human mapping information) MGD: Mouse Genome Database SGD: Saccharomyces Genome Database Funcional TRANSFAC: Transcription Factors Vector Database Organelle Genome Database GOBASE Tecnologia de Seqüenciamento EST: Expressed Sequence Tags GSS: Genome Survey Sequences STS: Sequence Tagged Sites HTG: High Throughput Sequences
 MGD: Mouse Genome Database. SGD: Saccharomyces Genome Database. Funcional. TRANSFAC: Transcription Factors. Vector Database. Organelle Genome Database GOBASE. Tecnologia de Seqüenciamento. EST: Expressed Sequence Tags. GSS: Genome Survey Sequences. STS: Sequence Tagged Sites. HTG: High Throughput Sequences.")
12
Tipos de Banco de Dados Nível de Curadoria Preliminar Arquivo
seqüências não terminadas - localizadas nos centros de seqüenciamento Arquivo repositório da informação redundante (várias seqüências do mesmo gene) submissor mantém controle editorial sobre registros Curadoria (cont.) Revisados (cont.)
 submissor mantém controle editorial sobre registros. Curadoria (cont.) Revisados (cont.)")
13
Tipos de Banco de Dados Nível de Curadoria Curadoria Revisadas
não-redundante cada registro pretende conter conhecimento corrente registro contém informações adicionais RefSeq: NCBI Database of Reference Sequences (mRNAs, proteínas, contigs genômicos e genomas/cromossomas completos) Swiss-Prot (seqüências de proteínas) EntrezGene (resumo de informações sobre loci genéticos em humanos, rato, camundongo, drosophila, levedura e zebrasfish) Clusters of Orthologous Groups (COGs) (sistema natural de família de genes de genomas completos) Revisadas cada registro revisado e comentado por especialistas ex, PROW: Protein Resources on the Web, revisões curtas sobre proteínas e famílias
 Swiss-Prot (seqüências de proteínas) EntrezGene (resumo de informações sobre loci genéticos em humanos, rato, camundongo, drosophila, levedura e zebrasfish) Clusters of Orthologous Groups (COGs) (sistema natural de família de genes de genomas completos) Revisadas. cada registro revisado e comentado por especialistas. ex, PROW: Protein Resources on the Web, revisões curtas sobre proteínas e famílias.")
16
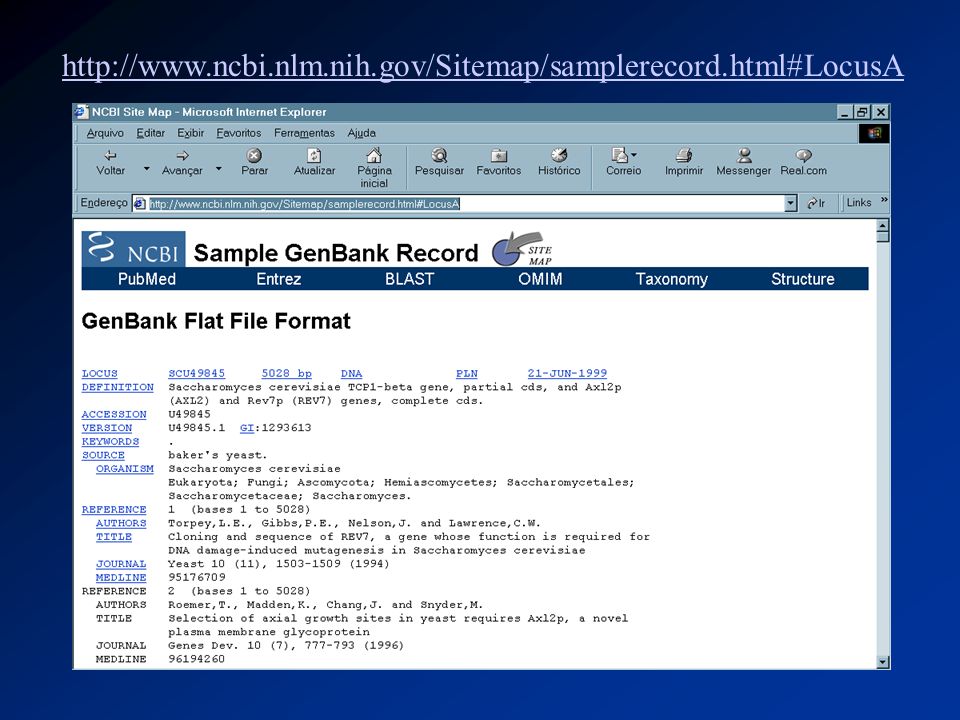
Estrutura do GenBank

18
Entrez

19
PubMed

20
TaxBrowser

21
BLAST

22
Busca em Banco de Dados Por texto - palavra chave
Entrez no NCBI Por seqüência: nucleotídeos ou amino ácidos (proteínas) BLAST no NCBI Basic Local Alignment Search Tool
 BLAST no NCBI. Basic Local Alignment Search Tool.")
26
Busca em Entrez Três níveis de complexidade de busca Básico
Enter: cystic fibrosis human Avançado (cont.) Complexo Boolean Enter: cystic fibrosis[titl] AND human[orgn]
 Complexo Boolean. Enter: cystic fibrosis[titl] AND human[orgn]")
27
Busca em Entrez Avançado Step 1: Select "Limits" option
Enter: cystic fibrosis Select "Title Word" as search field Press "Go" Step 2: Enter: human Select "Organism" as search field Step 3: Select "History" option Enter: #1 AND #2 (Note that Boolean operators must be in upper case. The OR and NOT operators are also available, and parentheses can be used to nest the search.)
")
28
Busca em BLAST Por seqüência de nucleotídeos ou de amino ácidos (proteínas) Comparação de seqüências para identificar similaridade significativa de DNA e PTN para inferir função, origem, filogenia Alinhamento: origem ou função comum Alinhamento Global ou Local (segmentos)
")
29
Busca em BLAST BLAST: Basic Local Alignment Search Tool
realiza comparações entre pares de seqüências buscando regiões com similaridade local NCBI BLAST WU-BLAST (Washington University) FASTA SSEARCH
 FASTA. SSEARCH.")
30
Busca em BLAST Similaridade x Homologia
identidade = ocorrência do exato mesmo nucleotídeo ou amino ácido na mesma posição nas seqüências alinhadas similaridade = considera combinações próximas e avaliada por medidas de diferença/igualdade homologia = dividem mesma ancestralidade com significado evolutivo Homologia => conceito central de Biologia

31
Busca em BLAST Algoritmos de BLAST, FASTA, SSEARCH
NÃO AVALIAM HOMOLOGIA MEDEM SIMILARIDADE E IDENTIDADE DE SEQÜÊNCIAS

32
Busca em BLAST Por que saber o quanto 2 seqüências são similares?
Porque a Natureza resolveu o mesmo problema várias vezes com significativa similaridade entre as soluções!

33
Busca em BLAST Alinhamento x Similaridade Requer meios de avaliar:
o que fazer primeiro? = Círculo vicioso Requer meios de avaliar: combinações perfeitas e imperfeitas (matches e mismatches) inclusão de gaps (intervalos) Matrizes ou Tabela de Valores: descrevem a probabilidade com sentido biológico de uma base ou amino ácido ocorrer num alinhamento
 inclusão de gaps (intervalos) Matrizes ou Tabela de Valores: descrevem a probabilidade com sentido biológico de uma base ou amino ácido ocorrer num alinhamento.")
34
Busca em BLAST Comparação de nucleotídeos -> por identidade
matches: tipicamente +1 ou +5 mismatches: tipicamente -1 ou -4 Comparação de amino ácidos -> mais complexo! função natureza química e freqüência matrizes de substituição penalidades maiores: menor funcionalidade freqüência de ocorrência

35
Busca em BLAST

36
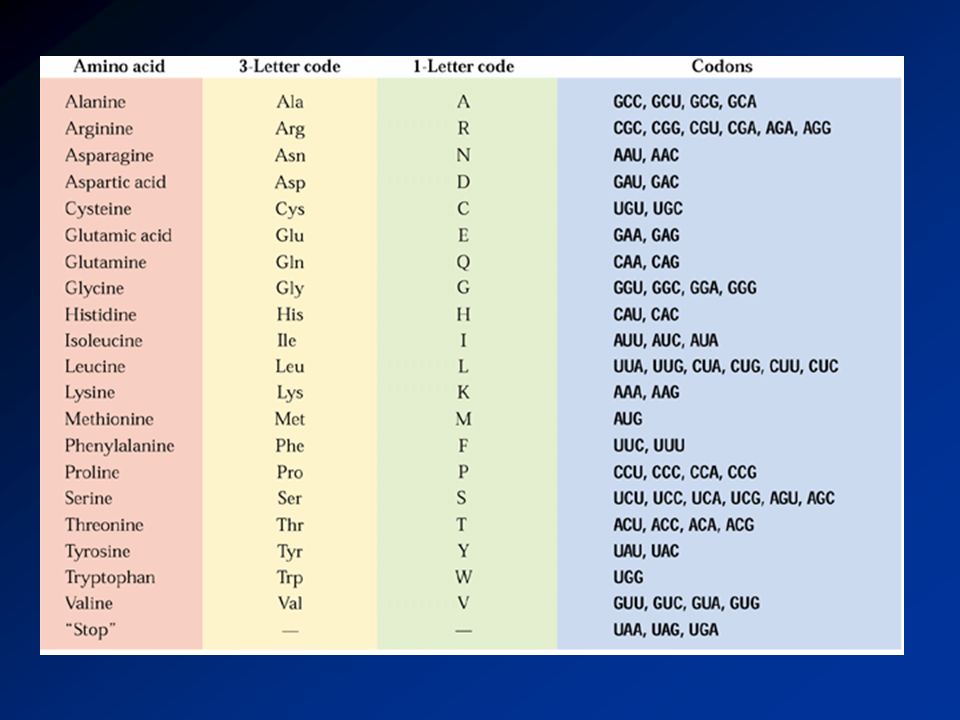
BLAST blastn DNA DNA DNA blastp PTN PTN PTN blastx DNA PTN PTN
Programa Seqüência Base Comparação blastn DNA DNA DNA blastp PTN PTN PTN blastx DNA PTN PTN tblastn PTN DNA PTN tblastx DNA DNA PTN

37
BLAST tblastn blastn blastp blastx tblastx Nucleotide Protein Sequence
Em 6 quadros Nucleotide Sequence Protein Sequence Translated Protein Sequence tblastn blastn blastp blastx Nucleotide DB Protein DB tblastx Translated DB (contain amino acid sequences) Em 6 quadros
 Em 6 quadros.")
38
Busca em BLAST Matrizes de Substituição:
baseadas em alinhamentos de alta confiança de diversas proteínas homólogas, avaliando freqüência de todas as substituições Point Accepted Mutation - PAM Blocks substitution - BLOSUM

39
Point Accepted Mutation - PAM
calculada baseada num modelo de distância evolucionária do alinhamento de seqüências próximas (mín. 85% idênticas) 34 super-famílias agrupadas em 71 árvores filogenéticas com substituições substituições separadas por tipo, normalizadas por freqüência de uso e convertidas para log dos odds Matriz PAM1 = prob. mudança de 1 aa em 100 Multiplicando a matriz por ela mesmo - matriz com graus arbitrários de proximidade
 34 super-famílias agrupadas em 71 árvores filogenéticas com substituições. substituições separadas por tipo, normalizadas por freqüência de uso e convertidas para log dos odds. Matriz PAM1 = prob. mudança de 1 aa em 100. Multiplicando a matriz por ela mesmo - matriz com graus arbitrários de proximidade.")
40
Point Accepted Mutation - PAM250

41
Blocks Substitution - BLOSUM
seqüências selecionadas para evitar aquelas muito relacionadas de ocorrência freqüente banco de dados - BLOCKS 2.000 blocos de segmentos de seqüência sem gaps alinhadas, caracterizando > de 500 grupos de famílias de ptns próximas seqüências dos blocos agrupadas freqüência de susbtituições entre grupos na família estimadas - prob. substituição importante nível de cut-off de % de identidade de seqüência que define os grupos (clusters) - ex. BLOSUM62
 - ex. BLOSUM62.")
42
Blocks Substitution - BLOSUM62

43
Busca em BLAST Matrizes de Substituição do BLAST-NCBI
Point Accepted Mutation - PAM PAM30, PAM70 Blocks substitution - BLOSUM BLOSUM45, BLOSUM62, BLOSUM80

44
Busca em BLAST Penalidades por Intervalo (gaps)
prevê inserções e deleções penalidades - limitar introdução mas flexível para extender (ocorre vários resíduos) BLAST-NCBI penalidades existência: 7, 8 e , 11 e 12 extensão:
 BLAST-NCBI. penalidades. existência: 7, 8 e 9 10, 11 e 12. extensão:")
45
Busca em BLAST Programação Dinâmica
solução ótima de busca de alinhamentos algoritmo de Smith-Waterman alinhamento local ao invés de global -mais relevante biologicamente

46
Busca em BLAST Heurística: Sensibilidade x Rapidez
FASTA: usa palavras exatas (strings) BLAST: usa matriz de scores (BLOSUM62) usa como semente de alinhamento com alto score realiza filtragem de seqüências repetidas (ex. CA) nos primeiros passos, gaps não são permitidos
 BLAST: usa matriz de scores (BLOSUM62) usa como semente de alinhamento com alto score. realiza filtragem de seqüências repetidas (ex. CA) nos primeiros passos, gaps não são permitidos.")
47
(a) 1. Filtra regiões de baixa complexidade 2. Gera lista de seqüências pequenas 11 bases nt ou 3 aa 3. Determina pares de maior score baseado na matriz e sem gaps 4. Reduz lista por limite estabelecido (b) 5. Busca no banco de dados combinações perfeitas para a lista gerada 6. Procura extender o alinhamento a partir dessa “semente” (c) 7. Tenta extender alinhamento até o score continuar a crescer 8. Gaps são incluídos 9. Determina significância de cada score calculando valor E.
 5. Busca no banco de dados combinações. perfeitas para a lista gerada. 6. Procura extender o alinhamento a. partir dessa semente (c) 7. Tenta extender alinhamento até o. score continuar a crescer. 8. Gaps são incluídos. 9. Determina significância de cada score. calculando valor E.")
48
BLAST Estatísticas: raw score: soma de scores de substituições e gaps (função das matrizes e não comparável) bit scores: versão em escala do score valor E: representa o número de alinhamentos esperados, equivalentes ou melhores do que o obtido numa busca contra um banco de dados aleatório com a mesma composição (ao acaso)
")
49
BLAST Submissão de seqüência - query
Alinhamento no banco de dados - target Melhores de escores são relatados usar valor E valor E <0.01 Submissão: DNA, PTN ou DNA como PTN Proteínas: melhor para identificar homologias DNA: degeneração do terceira base do códon

52
Blastn

53
Blastp

54
BLAST Interpretação de função: cautelosa!
Similaridada restrita a domínios Área de cobertura do alinhamento Erros de anotação no banco de dados Falta de homologia - distanciamento evolutivo

55
Alinhamento Múltiplo Alinhamento de 3 ou mais seqüências, com inserção de espaços (gaps) para que resíduos com posição estrutural e/ou co-ancestralidade comuns estejam alinhados na mesma coluna. Algoritmo: ClustalW
 para que resíduos com posição estrutural e/ou co-ancestralidade comuns estejam alinhados na mesma coluna. Algoritmo: ClustalW.")
Apresentações semelhantes