Carregar apresentação
A apresentação está carregando. Por favor, espere

1
AS HEMOGLOBINOPATIAS JULIANA ZIMMERMMANN

2
A ANEMIA FALCIFORME FREQUENCIA:
É uma das doenças hereditárias mais comuns no Brasil. Ela afeta principalmente a população negra, mas pode acometer qualquer raça. Aproximadamente 1 criança afro-brasileira em cada crianças nascem com a doença falciforme. Cerca de 1 em cada 8 afro-brasileiros tem o que é chamado de traço falcêmico.

3
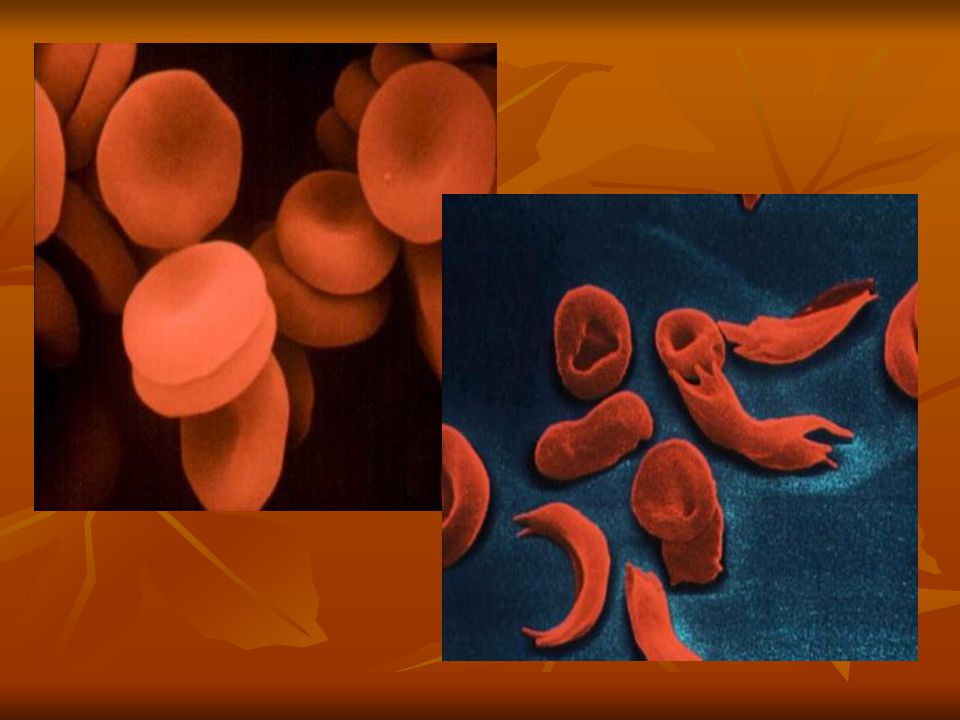
CONCEITO Trata-se de uma doença caracterizada pela troca do aminoácido ácido glutâmico pelo amnioácido valina. Esta substituição de aminoácidos é a responsável pela alteração na hemoglobina, determinando o processo de afoiçamento.

5
HEMOGLOBINA NORMAL E A FALCÊMICA
A hemoglobina normal, chamada de hemoglobina A é sempre feita de duas cadeias alfa globínicas e duas cadeias beta globínicas. A hemácia normal contém cerca de 95% de hemoglobina A. A hemoglobina, como toda proteína contém uma fileira de aminoácidos.

6
A HEMOGLOBINA NORMAL Na hemoglobina adulta normal as cadeias de globina são designadas alfa e beta. As quatro cadeias são dobradas e acomodadas juntas para formar um tetrâmero globular. As cadeias a e b são codificadas por genes em lóci distintos: Lócus alfa ; no cromossomo 16 Lócus beta ; no cromossomo 11

7
TIPOS DE HEMOGLOBINA A Hb A ( 97% das hemoglobinas ) possui 2 cadeias alfa (141 AA) e duas cadeias beta (146 AA). Essas cadeias possuem a propriedade de liberação e fixação do oxigênio. Por isso os outros tipos de Hb não transportam efetivamente o oxigênio. Hemoglobina F - formada por 2 cadeias alfa e duas cadeias gama.( 1% das Hb) - É a prevalente nos recém natos até 6 meses de vida.
 possui 2 cadeias alfa (141 AA) e duas cadeias beta (146 AA). Essas cadeias possuem a propriedade de liberação e fixação do oxigênio. Por isso os outros tipos de Hb não transportam efetivamente o oxigênio. Hemoglobina F - formada por 2 cadeias alfa e duas cadeias gama.( 1% das Hb) - É a prevalente nos recém natos até 6 meses de vida.")
8
Hemoglobina S - É mutante, ocorre pela troca do ac
Hemoglobina S - É mutante, ocorre pela troca do ac. glutâmico pela valina ( no sexto AA da cadeia b da HbA.

9
A HEMOGLOBINA S Quando a pessoa recebe de um dos pais a hemoglobina A e de outro a hemoglobina S, ele é chamado de “traço falcêmico”, sendo representado por AS. O portador de traço falcêmico não é doente, sendo portanto, geralmente assintomático e só é descoberto quando é realizado um estudo familiar.

10
Na Doença Falciforme as hemácias contém uma hemoglobina que é um pouco diferente da hemoglobina normal. Nas hemácias dos pacientes com Anemia Falciforme 90% das hemoglobinas são hemoglobinas S.

11
OS CRUZAMENTOS

16
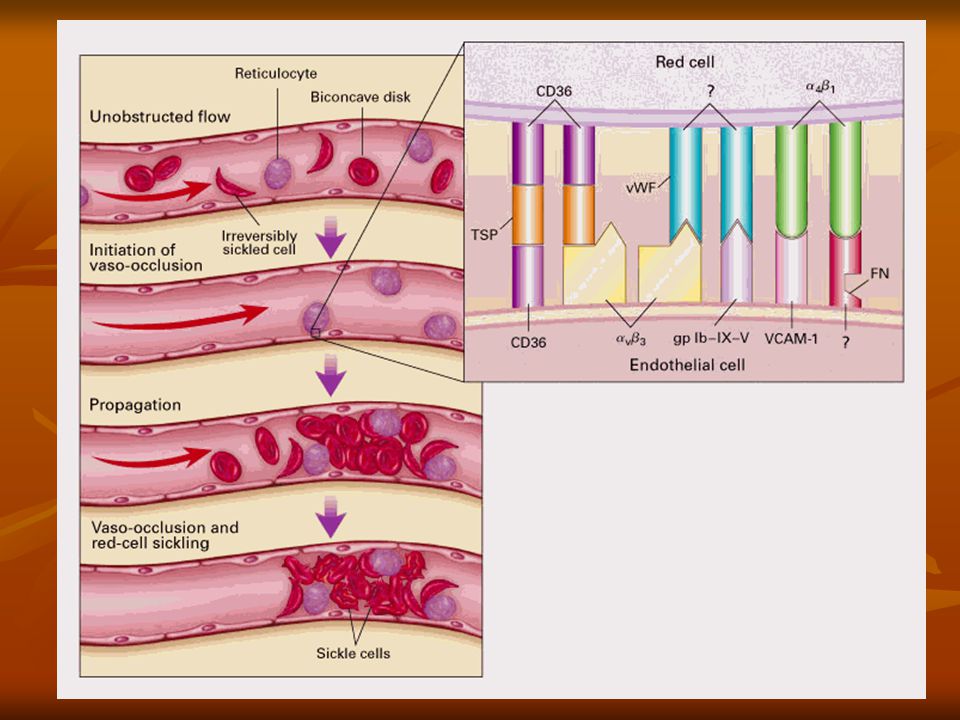
FISIOPATOLOGIA Diferentes situações podem levar as células com hemoglobina S a se afoiçarem, como, por exemplo, hipóxia, desidratação e febre.

17
FISIOPATOLOGIA

19
ASPECTOS CLÍNICOS 1. Crise de Dor
É o sintoma mais freqüente. A dor pode se localizar nos ossos ou nas articulações, no tórax, no abdômen, podendo atingir qualquer local do corpo (desidratação, stress, emocional) 2. Icterícia (cor amarela nos olhos) É o sinal mais freqüente da doença. Quando o glóbulo vermelho se rompe, libera bilirrubina que se impregna nas mucosas e é eliminada na urina (urina cor de coca-cola)
 2. Icterícia (cor amarela nos olhos) É o sinal mais freqüente da doença. Quando o glóbulo vermelho se rompe, libera bilirrubina que se impregna nas mucosas e é eliminada na urina (urina cor de coca-cola)")
20
3. Síndrome Mão-pé Nas crianças pequenas as crises de dor podem ocorrer nos pequenos vasos das mãos e dos pés causando inchaço, dor e vermelhidão no local. 4. Infecções Podem ocorrer infecções freqüentes localizadas na garganta, pulmões e ossos. Estas infecções devem ser vistas pelo médico hematologista tão logo apareçam, podem ser muito graves e até fatais. Todos os pacientes devem estar com a caderneta de vacinação atualizada.

21
5. Úlceras de Perna Úlceras de perna ocorrem, freqüentemente, próximo aos tornozelos 6. Seqüestro do Sangue no Baço O afoiçamento dos glóbulos no baço pode levar ao seqüestro do sangue. Há palidez e dor no baço e é uma emergência.

22
TRATAMENTO Profilaxia Ingesta de líquidos Hidratação venosa
Antibióticos: infecção

23
TALASSEMIAS Trata-se de um defeito na síntese das cadeias protéicas da hemoglobina. Prevalência: Talassemia deriva da combinação das palavras gregas thalassa = mar, e emas =sangue. Com esta palavra, os médicos queriam descrever uma doença do sangue cuja origem está nos países banhados pelo mar, e mais precisamente no Mediterrâneo, como Itália e Grécia. Hoje a doença se alastrou praticamente no mundo todo. Percentagens relevantes de portadores de Talassemia são registrados no Canadá, Estados Unidos, Brasil

24
TIPOS DE TALASSEMIA TALASSEMIA ALFA
As a Talassemias são anemias microcíticas hereditárias devido à redução da síntese de cadeias polipeptídicas alfa. A maioria das Talassemias Alfa são decorrentes de deleção de gene. Cada pessoa normal herda quatro gens globínicos afa , podendo-se escrever o genótipo assim: a a /a a .A deleção de um ou mais genes X globínicos determinará a gravidade da x talassemia.

25
TIPOS DE TALASSEMIA ALFA
Carreador Silencioso (portador silencioso): Ocorre devido a deleção de um gen alfa. Não existem manifestações hematológicas. As hemácias não são microcíticas; Há uma leve diminuição da síntese de cadeias alfa Traço alfa Talassêmico (talassemia alfa heterozigótica): Caracteriza-se por uma anemia hipocromica microcítica leve, devido à mutação ou deleção de dois gens alfa. Os níveis de HbA2 estão baixos ou na faixa inferior da normalidade. Cerca de 2% a 3% da população negra tem este traço .
: Ocorre devido a deleção de um gen alfa. Não existem manifestações hematológicas. As hemácias não são microcíticas; Há uma leve diminuição da síntese de cadeias alfa. Traço alfa Talassêmico (talassemia alfa heterozigótica): Caracteriza-se por uma anemia hipocromica microcítica leve, devido à mutação ou deleção de dois gens alfa. Os níveis de HbA2 estão baixos ou na faixa inferior da normalidade. Cerca de 2% a 3% da população negra tem este traço .")
26
Doença da Hb H: É uma doença moderadamente grave, com alterações ósseas e esplenomegalia. 3 genex x afetados Hidropsia Fetal / síndrome de Hb de Bart: Esta forma mais grave das alfa Talassemias está correlacionada com a ausência total da síntese de cadeias alfa . Os fetos afetados geralmente são prematuros e natimortos ou morrem logo após o nascimento. Esta síndrome ocorre principalmente em chineses, porém a migração de pessoas do sudeste da Ásia torna esta condição mais relevante no nosso meio.

27
TALASSEMIA BETA Mais comum em nossa região
Associada a diminuição da síntese das cadeias B Clínica: Talassemia menor (minor) Em geral o paciente, embora assintomático, manifesta o desconforto do cansaço e dores nas pernas com relativa freqüência. O eritrograma revela anemia microcítica e hipocrômica.
 Em geral o paciente, embora assintomático, manifesta o desconforto do cansaço e dores nas pernas com relativa freqüência. O eritrograma revela anemia microcítica e hipocrômica.")
28
Talassemia maior: O paciente com talassemia beta maior requer a necessidade de contínuas transfusões de sangue, medicações e vacinações específicas, além do diagnóstico precoce. Quando todos esses requisitos são obedecidos, a talassemia beta maior evolui moderadamente bem até a idade adulta.

29
TALASSEMIA BETA : origem genética
Quando o bloqueio do gene beta é total e afeta apenas um dos dois genes da pessoa (b/-), o portador é caracterizado geneticamente como talassemia beta heterozigoto, ou clinicamente como talassemia beta menor. Por outro lado, a lesão total de ambos os genes beta causa a talassemia beta homozigota ou talassemia beta maior.
, o portador é caracterizado geneticamente como talassemia beta heterozigoto, ou clinicamente como talassemia beta menor. Por outro lado, a lesão total de ambos os genes beta causa a talassemia beta homozigota ou talassemia beta maior.")
Apresentações semelhantes