Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Descobrindo genes de doenças humanas
Carolina Magalhães Drielly Braite Fernanda Rodrigues Fernanda Valiati Mariana Brutschin Profª Fabiana Seixas, Dra.

2
Histórico 1856 - Descoberta da Hereditariedade
Genética baseada no melhoramento genético Século XX

3
Genética do desenvolvimento Genética comportamental
Histórico – Século XX Genética molecular Citogenética Genética do desenvolvimento Genética do câncer Genética comportamental Surgimento de novas técnicas de estudo genético

4
Histórico 2003 – Projeto Genoma Humano Atualidades
Reconhecimento dos fatores genéticos na etiologia das doenças Genética clínica Área Promissora Atualidades Imprintings Genômicos: As contribuições dos genomas materno e paterno podem variar e acarretar em doenças com fenótipos diferentes.

5
Por que estudar os genes?
Diagnósticos de doenças Tratar, prevenir ou curar doenças genéticas Função e manifestação desses genes Mecanismos de hereditariedade Aconselhamento genético Banco de dados

6
Genômica Genômica Estrutural Genômica Funcional
Ramo da genética que estuda a natureza física e o funcionamento do material genético contido no conjunto de cromossomos de cada espécie Genômica Estrutural Determina a posição cromossômica e a sequência de bases dos genes de diversas espécies, incluindo a humana. Contribui para a detecção de mutação nos genes humanos. Genômica Funcional Descreve as funções dos genes Analisa como os genes estão sendo expressos ou silenciados.

7
Aplicações da genômica
Desenvolvimento de testes para a detecção de mutações gênicas Exemplo: Mutações nos genes BRCA1 e BRCA2, que determinam as variantes hereditárias dos cânceres de mama e ovário. SuscePtibilidade a doenças!

8
Baseada em testes genéticos e análises moleculares que
Saúde MEDICINA Doença MEDICINA GENÔMICA PERSONALIZADA Baseada em testes genéticos e análises moleculares que permitem o conhecimento do mapa de predisposições genéticas de cada indivíduo.

9
Genome-Wide Association Studies (GWAS)
Rápido escaneamento de marcadores em genomas de uma série de pessoas a fim de encontrar variações genéticas associadas com uma doença em particular. Bases para a Medicina Genômica Personalizada

10
GWAS Escaneamento em equipamentos automatizados
Dois grupos de participantes Portadores da doença Saudáveis Obtenção do DNA genômico Amostras de sangue ou esfregaço bucal Deposição em chips de DNA Escaneamento em equipamentos automatizados

11
Variações “associadas” com a doença
GWAS Levantamento do genoma de cada participante Busca por variações SNPs Variações com maior frequência em portadores Variações “associadas” com a doença

12
Variações “associadas” podem não causar diretamente a doença
GWAS Variações “associadas” podem não causar diretamente a doença É necessário o sequenciamento de DNA da região do genoma em particular, a fim de identificar a exata modificação genética envolvida na doença.

14
Published Genome-Wide Associations through 12/2010,
1212 published GWA at p<5x10-8 for 210 traits NHGRI GWA Catalog

17
Doenças Neurodegenerativas
Perda excessiva dos neurônios Deterioração do tecido nervoso na substância cinzenta do SNC Neurônios são células do SN responsáveis pelo impulso elétrico

18
Identificação quanto a causa – esporádica/incidência familiar.
Diagnóstico Identificação quanto a causa – esporádica/incidência familiar. Diagnóstico Molecular Avaliação da estrutura dos componentes do DNA. Investigação Molecular Identificação das alterações dos genes ou marcadores biológicos suscetíveis a doença. Patologia Contribuição para o estudo. Alterações: Medula espinhal, músculos e nervos.

19
Genes envolvidos com Doenças Neurodegenerativas

20
Afeta prncipalmente IDOSOS
Doença de Alzheimer Doença neurodegenerativa Afeta prncipalmente IDOSOS Progressiva Irreversível

21
Manifestações Clínicas:
Perturbações de memória Declínio cognitivo Distúrbios comportamentais Perda de autonomia Pensamento abstrato

22
Grande problema de saúde pública!
IDADE Fatores ambientais Eventos genéticos Afeta: cerca de 13% dos indivíduos com 65 anos ou mais 30 -50% dos indivíduos com 80 anos ou mais Grande problema de saúde pública!

23
Neuropatologia: Placas amiloides, compostas por peptídeo amiloide- β (Aβ) Emaranhados de neurofibrilas contendo proteína Tau hiperfosforilada. Microscópica neuropatologia da doença de Alzheimer, mostrando uma placa neuríticas (canto inferior esquerdo canto) eemaranhados neurofibrilares (canto superior direito).
 eemaranhados neurofibrilares (canto superior direito).")
24
Genes associados à Doença de Alzheimer
DA de início precoce DA de início tardio APP PSEN1 ApoE PSEN2 5% do total dos casos de DA 95% do total dos casos de DA

25
APP (proteína precursora de amiloide)
Localização : 21q21.2 Identificação a partir da purificação dos depósitos de peptídeo amiloide da placa neural. Apresenta 695 resíduos de AA, composto por 18 éxons.

26
APP Peptídeo amilóde Aβ - 42 Peptídeo amilóde Aβ - 40 SECRETASES!

27
APP é uma proteína trans-membrana
Sua degradação anormal produz fragmentos peptídeos Aβ42 que se agregam e formam a placa neural

28
maior produção do peptídeo Aβ
Mutações Mais de 32 mutações missense (não silenciosa) Sítios de clivagem das secretases Domínio trans-membrana nos éxons 16 e 17 V717I substituição do aminoacido valina pelo isoleucina na posição 717 da proteína V717I maior produção do peptídeo Aβ produção na forma Aβ42
 Sítios de clivagem das secretases. Domínio trans-membrana nos éxons 16 e 17. V717I substituição do aminoacido valina pelo isoleucina na posição 717 da proteína. V717I. maior produção do peptídeo Aβ. produção na forma Aβ42.")
29
PSEN1 (Presensilina 1) Localização : 14q24.3
Apresenta 12 éxons e codifica para uma proteína com 467 aminoácidos. São responsáveis pela clivagem da APP a partir da γ-secretase.

30
Mutações Mais de 176 mutações na PSEN1 - maioria missense. Aβ42 / Aβ40
L166P Adolescência Elevada produção de Aβ42 L166P mudança na região 166 do AA leucina para o AA prolina Defeitos em PSEN1 : formas mais graves de DA penetrância completa início logo aos 30 anos de idade

31
PSEN2 (Presensilina 2) Localização : 1q42.13
Possui 12 éxons que codificam para um peptídeo de 448 aminoácidos.

32
proporção de Aβ42 sobre Aβ40
Mutações Apenas 14 mutações foram relatadas I141N Perda do éxon 5 Aumento da proporção de Aβ42 sobre Aβ40 I141N na substituição de uma isoleucina por uma asparagina nos resíduos 141

33
ApoE (Apolipoproteína E)
Localização : 19q13.2 Composto por 4 éxons que codificam uma proteína de 299 aminoácidos. No éxon 4 há a presença de três isoformas proteicas ε2 (CIS/CIS); ε3 (CIS/ARG); ε4 (ARG/ARG)
; ε3 (CIS/ARG); ε4 (ARG/ARG)")
34
Risco de desenvolver DA até os 85 anos:
4/ 4 55% 3/ 4 27% 3/ 3 9% ApoE principal fator de risco para desenvolvimento da doença, representado em excesso nos indivíduos com DA. A herança de um ou dois desses alelos eleva até 5x a probabilidade de desenvolvimento da doença!

35
A apo E vem sendo associada à doença de Alzheimer desde 1993

36
Descobertas recentes:

37
GWAS - genome-wide association study
Procura por SNPs Genoma de indivíduos portadores da doença Genoma de indivíduos normais Consórcio genético da doença de Alzheimer (ADGC) reuniu banco de dados de 9 coortes a partir dos 29 Institutos Nacionais do Envelhecimento (NIA)
 reuniu banco de dados de 9 coortes a partir dos 29 Institutos Nacionais do Envelhecimento (NIA)")
38
Nesse estudo foram identificados os genes MS4A4A, CD2AP, EPHA1, CD33 que são fatores de risco para a DA e comprovada a associação de genes anteriormente descobertos. Até o momento foram confirmados então 10 genes envolvidos com a DA de início tardio (ApoE, CR1, CLU, PICALM, BIN1, EPHA1, MS4A, CD33, CD2AP e ABCA7)
")
39
Diagnóstico Molecular

40
GENOTIPAGEM de ApoE A metodologia de escolha para se determinar o polimorfismo da apo E é a genotipagem. A técnica envolve uma reação em cadeia da polimerase (PCR) e um estudo do poliformismo do tamanho dos fragmentos de restrição (RFLP). Uma PCR é realizada sobre o DNA genômico. O fragmento apo E amplificado é então digerido com uma de restrição, que diferencia os alelos alternativos.
 e um estudo do poliformismo do tamanho dos fragmentos de restrição (RFLP). Uma PCR é realizada sobre o DNA genômico. O fragmento apo E amplificado é então digerido com uma de restrição, que diferencia os alelos alternativos.")
41
Terapia Gênica Um dos potenciais terapêuticos para o tratamento da DA seria a diminuição da produção ou o aumento da degradação do peptídeo beta-amiloide. Grande potencial terapêutico Monócitos foram modificados geneticamente para expressar a enzima neprilisina (NEP) que é capaz de degradar esse peptídeo beta-amiloide.
 que é capaz de degradar esse peptídeo beta-amiloide.")
42
Doença de Huntington Doença neurodegenerativa hereditária
Autossômica dominante com penetrância Completa 5 a 10 casos por habitantes A doença de huntington é uma doença neurodegenerativa de herança autossômica dominante de penetrância completa. A incidência é de 5 a 10 casos a cada habitantes.

43
Manifestações Clínicas
Entre 35 e 50 anos depressão Demência Distúrbios psiquiátricos transtorno obsessivo-compulsivo Raciocínio Lento Deterioração progressiva das funções cognitivas As manifestações clínicas ocorrem normalmente entre os 35 e 50 anos. Os principais sintomas são demência, movimentos involuntários e anormais. Deterioração progressiva das funções cognitivas, como raciocínio lento, dificuldade de concentração, organização e decisão, perda de memória. E também distúrbios psiquiátricos como depressão, transtorno obsessivo compulsivo e ansiedade. ansiedade Dificuldade de concentração, organização, decisão Movimentos involuntários e anormais Perda de Memória

44
Corpo estriado Problemas com o movimento
Nesses cortes cerebrais podemos comparar as diferenças na estrutura, bem como a degeneração, em pacientes normais e pacientes com huntington. Na região marcada, podemos perceber a degeneração do corpo estriado, que está diretamente relacionada com os problemas no movimento. Nessa outra região, a do córtex, também vemos uma degeneração, e esta está relacionada com problemas emocionais, cognitivos e psiquiátricos. Corpo estriado Problemas com o movimento Córtex Problemas emocionais, cognitivos e psiquiátricos

45
Genes Associados Glutamina Mutação região 4p16.3
Gene IT15, extremidade 5’ repetições de trinucleotídeo Glutamina Essa doença é causada por uma mutação no Cromossomo 4, braço pequeno, região 1 da banda 6 da sub-banda 3, no gene IT15, EXTREMIDADE 5’, próximo a região telomerica. Essa mutação é causada por uma repetição do trinucleotídeo CITOSINA, ADENINA E GUANINA que codificam pro aminoácido Glutamina. É uma mutação instável, principalmente quando o pai afetado pois a espermatogenese é mais suscetível a erros. Quanto maior for o numero de poliglutaminas, mais cedo será a manifestação. A instabilidade da mutação causa um fenomeno de antecipação. Esse fenomeno é raro e ocorre com mutações que contém mais de 60 repetições. Mutação instável – pai afetado Número de Poliglutaminas Idade de manifestação Fenômeno de antecipação raro – repetições

46
Antecipação genética

47
Gene IT15 1983 – análise de ligação, sonda G8
Primeiro gene responsável por doença localizado em humanos Codifica huntingtina O gene IT15 foi identificado em 1983 através de análises de ligação utilizando a sonda G8. Possui grande importancia por ser o primeiro gene responsável por doença localizado em humanos Ele codifica para uma proteína conhecida como HUNTINGTINA.

48
Huntingtina Presente em vários tecidos do corpo Cérebro
Cérebro: quase exclusiva do citoplasma neuronal axônios, dendritos e corpo celular Possíveis funções na célula: Migração vesicular e de exocitose de substâncias neurotransmissores e enzimas Mecanismos anti-apoptóticos A huntingtina está presente em vários tecidos do corpo, principalmente no cérebro, onde ela é quase exclusiva do citoplasma neuronal, mas está presente também axônios, dendritos e corpo celular.

49
Alteração da função normal
Huntingtina Normal Huntingtina DH X 35 resíduos CAG +38 resíduos CAG Citoplasma Neuronal Núcleo Neuronal Alteração da função normal

50
Interações Celulares Proteína Mutante toxicidade Cito- Doença de Huntington

51
Expressão genes para transmissão neuronal
Hunting-tina Sp1 TAFII130 Hunting-tina Sp1 TAFII130 Genes não são expressos Huntingtina se liga a uma proteina SP1 que atua como fator de transcrição, ligando-se a TAFII130 Degradação e mudanças na função neuronal

52
Huntingtina DH Huntingtina Normal
Sequestra elementos repressores de transcrição Capacidade de reter elementos repressores é diminuída BDNF é transcrito normalmente BDNF e outros genes neuronais não são transcritos * Outros genes neuronais são transcritos normalmente Redução dos genes neuronais

53
Huntingtina DH + Proteínas do citoesqueleto
Huntingtina + Proteínas do citoesqueleto Interações mais fortes Permite circulação de vesículas nos microtúbulos Reduz o transporte de vesículas nos microtúbulos

54
Fortes interações com HAP-1 Morte Celular

55
Perda do suporte neurotrófico da BDNF do neurônio cortical para o estriado
Disfunção Mitocondrial Reciclagem Vesicular alterada Desregulação transcricional Agregados da proteína: citoplasma e núcleo Aumento da autofagia Disfunção e morte neuronal precoce Transporte Axonal Prejudicado DH

57
Tratamento Tratamento sintomático:
Depressão – fluoxetina, carbamazepina Paranóia e psicose – clozapina, carbamazepina Ansiedade – benzodiazepínicos Convulsões – clonazepam, ácido valpróico

58
Abordagens do Artigo Repressão da Huntingtina mutante através de RNAi
Atrasar ou Impedir início dos sintomas em DH RNAi induzido por RNAs hairpin curto (shRNAs) a fim de reduzir a expressão de htt mutante e melhorar as anormalidades associadas a DH em um modelo de camundongo transgênico da doença. A hipótese seria a repressão da... Nesse estudo, os pesquisadores...
 a fim de reduzir a expressão de htt mutante e melhorar as anormalidades associadas a DH em um modelo de camundongo transgênico da doença. A hipótese seria a repressão da... Nesse estudo, os pesquisadores...")
59
Terapias com RNAi possui potencial
RNAi direcionado contra o gene que codifica Huntingtina Reduz RNAm Melhora nas anormalidades comportamentais e neuropatológicas Reduz expressão da proteína em culturas de células e em células do cérebro do rato Terapias com RNAi possui potencial para o tratamento de DH

60
Esclerose Lateral Amiotrófica (ELA)
Doença de Lou Gehrig ou Doença de Charcot ELA Degeneração dos neurônios motores Sensibilidade mantida Movimentos voluntários dos músculos

61
Manifestações clínicas
Cãibras e tremores musculares Atrofia dos membros superiores e inferiores Perda de força muscular

62
Diagnóstico Eletroneuromiografia (EMG) Espirometria
Os exames mais comumente realizados são de sangue e urina, estudo do líquido cefalorraquiano ou liquor (o líquido que envolve o cérebro e a medula espinhal), eletroneuromiografia (EMG) (estudo da atividade elétrica dos nervos e dos músculos), espirometria (para avaliar a capacidade pulmonar) e ressonância magnética (estudo de imagem que permite ver o formato do cérebro e da medula espinhal). Ressonância Magnética Coleta do líquido cefalorraquidiano
, eletroneuromiografia (EMG) (estudo da atividade elétrica dos nervos e dos músculos), espirometria (para avaliar a capacidade pulmonar) e ressonância magnética (estudo de imagem que permite ver o formato do cérebro e da medula espinhal). Ressonância Magnética. Coleta do líquido cefalorraquidiano.")
64
Genes envolvidos com a ELA
Apenas 10% da doença tem caráter genético Principais genes estudados SOD1 FUS/TLS TARDBP 13 regiões do genoma denominadas ALS

65
Localização: braço longo do cromossomo 2
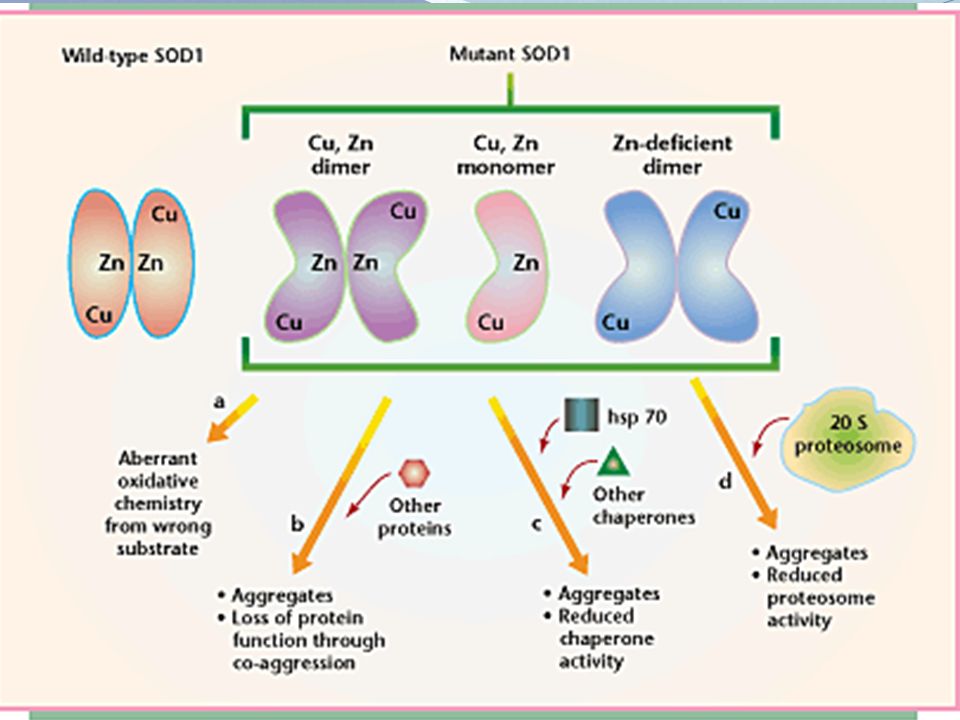
Região ALS1: SOD1 Função: Enzima Superóxido Disumutase - enzima antioxidante - conversão de íons superóxido em peróxido de hidrogênio Mutação: gera proteína com função altamente tóxica – incapacidade de dimerizar Cu e Zn Localização: braço longo do cromossomo 2 Mutações no gene SOD1 é a forma mais comum de esclerose lateral amiotrófica hereditária, responsável por 20% de todas as formas familiares ALS e correspondente a 1% -2% dos casos de ELA. A substituição da alanina-a-valina na posição 4 da SOD1 (SOD1A4V) é a mutação mais importante na América do Norte, responsável por 50% dos casos. Esse gene codifica a CuZn superóxido dismutase, enzima antioxidante que atua na conversão de íons superóxido em peróxido de hidrogênio. Mais de 140 mutações foram descritas no SOD1, sendo que elas alteram a estrutura da proteína, que passa a ter função tóxica no neurônio.
 é a mutação mais importante na América do Norte, responsável por 50% dos casos. Esse gene codifica a CuZn superóxido dismutase, enzima antioxidante que atua na conversão de íons superóxido em peróxido de hidrogênio. Mais de 140 mutações foram descritas no SOD1, sendo que elas alteram a estrutura da proteína, que passa a ter. função tóxica no neurônio.")
68
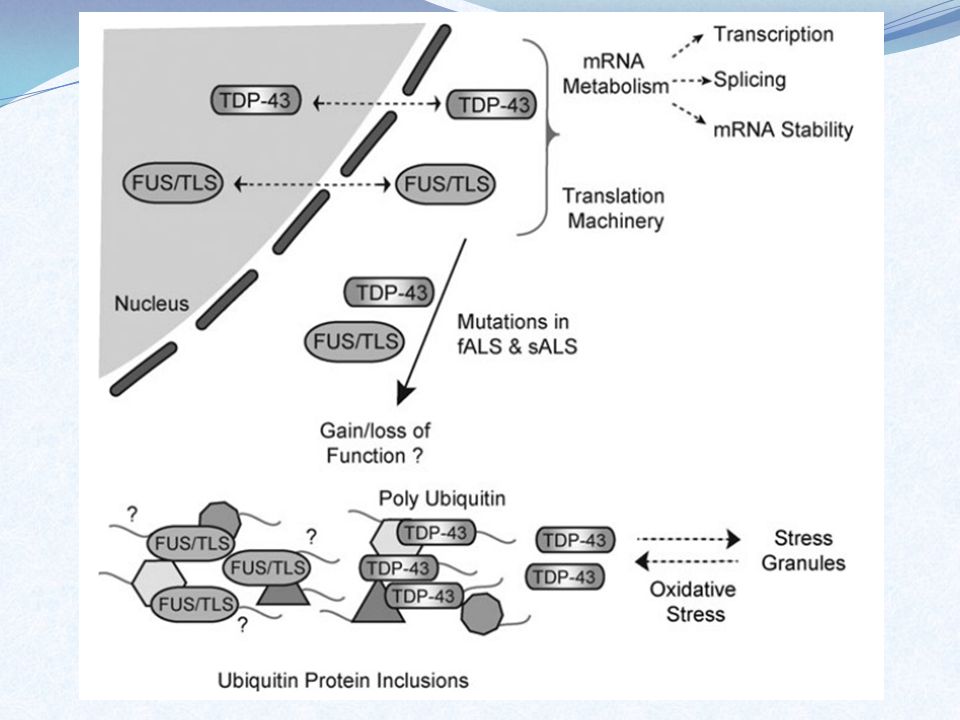
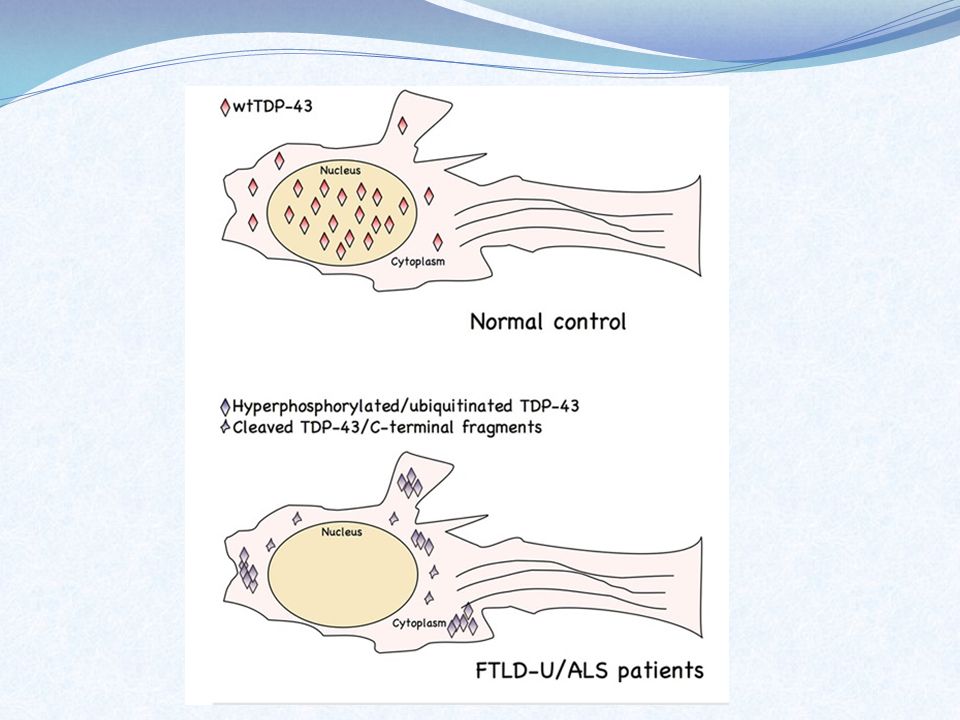
Região ALS6: FUS/TLS Função: regulação de como os RNAs mensageiros são criados, modificados e transportados para produzir as proteínas Mutação: proteína encontrada aglomerada fora do núcleo – efeito tóxico Localização: cromosssomo 16

71
Localização: braço curto do cromossomo 1
Região ALS10: TARDBP Função: produz proteína TDP43 relacionada a regulação da expressão gênica Mutação: aglomerados da proteína tóxicos ou ligada a grupos for a do núcleo – apoptose dos neurônios Localização: braço curto do cromossomo 1

75
Terapia Gênica Estudos realizados com camundongos transgênicos que expressam o gene SOD1 contendo a mutação G93A. Utilizou-se RNAi para impedir a tradução dos mRNA não ocorrendo a produção da proteína de conformação errada. Essa técninca utilizou como vetor os Lentivírus e obersevou-se como resultado um retardo do início dos sintomas da ELA.

76
Outros genes envolvidos…

77
Doença de Parkinson 2ª doença neurodegenerativa mais comum
Afeta principalmente idosos > 50 anos de idade Prevalência aos 65 anos

78
DISTÚRBIO DO SISTEMA MOTOR
Doença de Parkinson Crônica Progressiva Perda de neurônios dopaminérgicos da substância cinzenta do SNC. dopamina SINTOMAS MOTORES DISTÚRBIO DO SISTEMA MOTOR

79
Diagnóstico Tratamento Característica morfológica
Corpos de Lewy Tratamento Tratamento por controle dos sintomas

80
Manifestações Clínicas
Após perda de 60% dos neurônios e diminuição de 80% da produção de dopamina Tremor Rigidez dos membros e do tronco; Bradicinesia ou lentidão dos movimentos; Instabilidade postural Diminuição do equilíbrio e da coordenação.

81
Causas Ação de neurotoxinas ambientais Produção de radicais livres
Anormalidades mitocondriais Envelhecimento cerebral Predisposição genética IDIOPÁTICA

82
Susceptibilidade genética
Forma esporádica 95% dos casos Etiologia multifatorial Fatores genéticos + ambientais Forma familiar 5% a 10% dos casos Histórico familiar Fatores genéticos Autossômica dominante Autossômica recessiva

83
Genes envolvidos Alfa-sinucleína (SNCA) Parkin UCH-L1
PINK-1 DJ-1 LRRK2 GBA ATP13A2 Alfa-sinucleína (SNCA) Parkin UCH-L1
 Parkin. UCH-L1.")
84
SNCA PARK 1 – SNCA 4q21.3 Herança: autossômica dominante
Codifica para a alfa-sinucleína Proteína pré-sináptica 140 a.a Principal componente dos corpos de Lewy

85
SNCA Mutações: A53T e A30P Impedem degradação protéica pela alfa-sinucleína Produção de protofibrilas em células nervosas Ligação de protofibrilas em vesículas sinápticas Excesso de dopamina no citosol

86
SNCA MUTAÇÕES Agregados protéicos intracelulares (Corpos de Lewy) Células dopaminérgicas prejudicadas A presença de alfa-sinucleína, ubiquitina e subunidades proteassômicas nos corpos de Lewy sugerem o envolvimento da alfa-sinucleína na etiologia da disfunção do complexo ubiquitina-proteassoma na DP, o qual é essencial para a degradação de proteínas danificadas.

87
Parkin PARK 2 – Parkin 6q25.2-27 Mutações são comuns
50% dos casos de DP Herança: autossômica recessiva

88
Mutações 95 já identificadas 40 rearranjos de éxons 28 deleções
14 multiplicações 43 SNPs 12 inserções e deleções Mutações mais frequentes: deleção do éxon 2, 3 e 4 mutações nos éxons 2 (255/256delA) mutação pontual no éxon 7 (924C>T)
 mutação pontual no éxon 7 (924C>T)")
89
Parkin Codifica para a parkina (ubiquinona)
Envolvimento na via ubiquitina-proteassoma Presente nos corpos de Lewy Domínio RING UBIQUITINA –LIGASE E3

90
Ubiquitinação de proteínas danificadas Cadeias de poliubiquitina
Ubiquitina-ligase E3 Ubiquitinação de proteínas danificadas Cadeias de poliubiquitina Degradação de proteínas danificadas

91
DOENÇA DE PARKINSON MUTAÇÕES DISFUNÇÃO PROTÉICA
FALHAS NA VIA UBIQUITINA-PROTEASSOMA ACÚMULO DE PROTEÍNAS DANIFICADAS NEUROTOXICIDADE APOPTOSE DE CÉLULAS NEURAIS DOENÇA DE PARKINSON

92
UCH-L1 PARK 5 – UCH-L1 4p14 Herança: autossômica dominante
Codifica para a enzima hidrolase L1 ubiquitina carboxiterminal

93
UCH-L1 Hidrolase L1 ubiquitina carboxiterminal
Enzima deubiquitinizante Hidrolisa cadeias de poliubiquitina em ubiquitina monomérica - RECICLAGEM Novos processos de ubiquitinação de proteínas danificadas

94
DOENÇA DE PARKINSON MUTAÇÕES NEUROTOXICIDADE
DISFUNÇÃO DA ENZIMA FALHA NA RECICLAGEM DOS MONÔMEROS ATIVIDADE DA VIA UBIQUITINA PROTEASSOMA REDUZIDA NÃO DEGRADAÇÃO DE PROTEÍNAS DANIFICADAS ACÚMULO DE PROTEÍNAS NEUROTOXICIDADE APOPTOSE DOENÇA DE PARKINSON

95
Outros genes envolvidos
Locus Localização cromossômica Gene Padrão de herança PARK 1 4q21.3 Alfa-sinucleína AD PARK 2 6q Parkin AR PARK 3 2p13 - PARK 4 4p15 PARK 5 4p14 UCH-L1 PARK 6 1p35-p36 PINK-1 PARK 7 1p36 DJ-1 PARK 8 12p11.2-q13.1 LRRK2 PARK 9 ATP13A2 PARK 10 1p32 PARK 11 2q36-q37

96
5 novos genes

97
5 novos genes Meta-análise de sequências variantes de cinco estudos GWAS da doença de Parkinson dos EUA e Europa Objetivos: investigar as associações de loci previamente identificados e identificar novos loci de risco para a doença avaliar as consequências biológicas de variantes de risco identificadas, como SNPs Examinar a associação de alelos variantes, tanto com a expressão gênica quanto com a metilação do DNA.

98
Diagnóstico molecular
Heterogeneidade de genes envolvidos Escassez de informações sobre mecanismos moleculares Investigação molecular Busca de marcadores de susceptibilidade É recomendada a realização de testes genéticos para a identificação de mutações no gene parkin em pacientes nos quais a doença tiver início antes dos 30 anos de idade.

99
Terapia gênica

100
Bibliografia Tobin A.J; Signer, E.R. Huntington’s disease: the challenge for cell biologists. Trends Cell Biol Dec;10(12):531-6. Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet Mar;20(3): Nasir, J; Goldberg ,Y.P; Hayden M.R. Huntington disease: new insights into the relationship between CAG expansion and disease. Hum Mol Genet. 1996;5 Spec No: Lynn M. ; Genetics of Alzheimer Disease; J Geriatr Psychiatry Neurol December ; 23(4): 213–227. doi: / Bird, T. ; Genetic Aspects of Alzheimer Disease; Genet Med April ; 10(4): 231– 239. doi: /GIM.0b013e31816b64dc.
: Nasir, J; Goldberg ,Y.P; Hayden M.R. Huntington disease: new insights into the relationship between CAG expansion and disease. Hum Mol Genet. 1996;5 Spec No: Lynn M. ; Genetics of Alzheimer Disease; J Geriatr Psychiatry Neurol December ; 23(4): 213–227. doi: / Bird, T. ; Genetic Aspects of Alzheimer Disease; Genet Med April ; 10(4): 231– 239. doi: /GIM.0b013e31816b64dc.")
101
Obrigado!

Apresentações semelhantes