Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Genômica/Bioinformática
BIOLOGIA/MEDICINA A Revolução da Genômica/Bioinformática

2
A Verdadeira Revolução
Início do séc. 20: Mendel e as leis da hereditariedade. 1944: DNA como elemento carreador da informação genética (Avery) 1953: Watson/Crick e aestrutura do DNA. Anos 70 e 80: Biologia Molecular/Biotecnologia Anos 90 e séc. 21: Genômica/Bioinformática
 1953: Watson/Crick e aestrutura do DNA. Anos 70 e 80: Biologia Molecular/Biotecnologia. Anos 90 e séc. 21: Genômica/Bioinformática.")
3
História da Biologia Molecular
1951 Fred Sanger, Amino Acid Sequence of Insulin 1953 Watson/Crick, Estrutura do DNA 1957 Francis Crick, Central Dogma, DNA RNA Protein 1960’s Nirenberg, Matthaei, The Genetic Code 1967 Shapiro and Beckwith, First gene cloned, LacZ 1972 Paul Berg, First recombinant DNA molecule 1973 Cohen/Boyer, First recombinant organism 1977 Maxam/Gilbert and Fred Sanger, DNA sequencing 1977 Fred Sanger, Complete sequence of phage X174 1978 David Botstein, Restriction Fragment Length Polymorphisms (RFLP) 1980 Kerry Mullis, PCR 1983 Lee Hood, First Automated DNA Sequencer
 1980 Kerry Mullis, PCR Lee Hood, First Automated DNA Sequencer.")
4
Sequenciamento do DNA Sanger, Gilbert (Nobel 1980)
")
5
Sequenciamento Automático
Leroy Hood 30kb por corrida

6
A era genômica

7
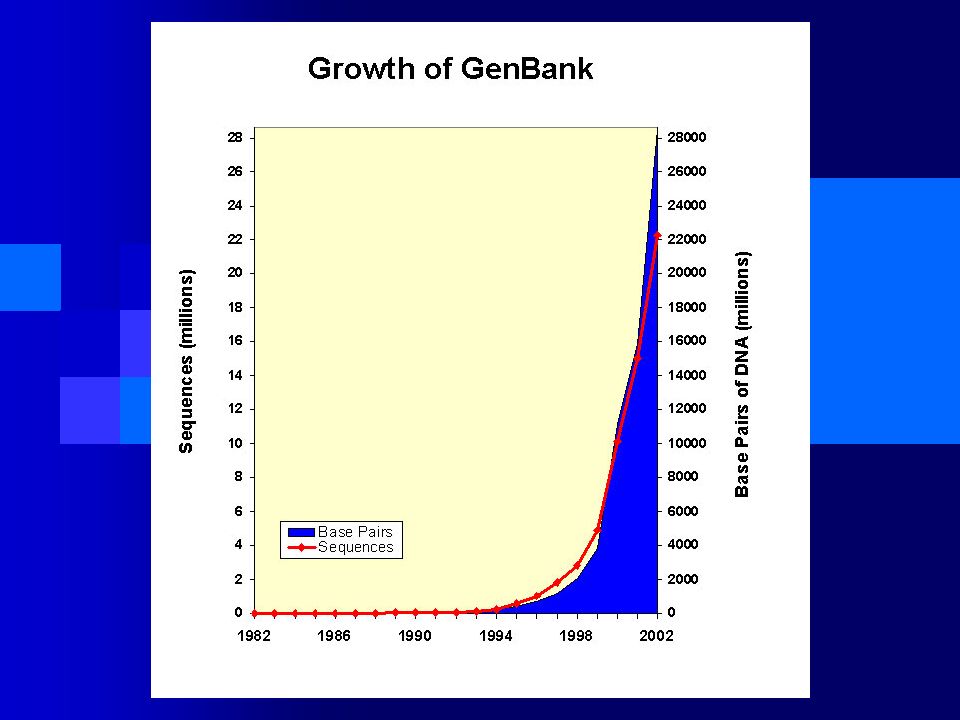
376 Genomas Concluídos! 251 como rascunho! 342 em andamento
327 Bacterial, 27 Archeal, 22 Eukaryotic 1995, Haemophilus influenzae 1996, Methanococcus jannaschii 1997, Saccharomyces cerevisiae 1997, Escherichia coli 1998, Caenorhabditis elegans 2000, Drosophila melanogaster 2000, Arabidopsis thaliana 2001, Homo sapiens 2002, Schizosaccharomyces pombe 2002, Oryza sativa 2002, Mus musculus 2005, Pan troglodites 09/07/2006

8
Um modelo genético

9
Genoma Humano 2001 International Consortium Celera Genomics
Grupos acadêmicos Celera Genomics Companhia Privada

10
A nova revolução da Genômica
454 Solexa - Illumina SOLiD - ABI ~120 MB de DNA por corrida ~01 GB de DNA por corrida ~03 GB de DNA por corrida 12KB/US$ KB/US$ 300KB/US$ Tecnologia de Capilar = 0.5KB/US$

11
Nova tecnologia Dispensa clonagem dos fragmentos em sistemas bacterianos Dispensa a preparação de DNA molde para sequenciamento Reações feitas em paralelo em volume extremamente pequeno - nanotecnologia

12
Aplicações Sequenciamento de Genomas Sequenciamento de Transcriptomas
sequenciamento de novo re-sequenciamento - variabilidade SNPs e mutações Sequenciamento de Transcriptomas variabilidade - splicing, poliadenilação quantificação de expressão gênica

13
Sequenciamento de novo

14
Re-sequenciamento

15
Transcriptoma

16
Projeto 454 Participantes: LICR-SP, LICR-NY, Venter Institute
Objetivo: Sequenciamento extensivo dos genes expressos na linhagem celular HCC1954 (tumor de mama) buscando conhecer, com um único set de dados, alterações genéticas e epi-genéticas neste tipo de câncer. Sequenciador: 454
 buscando conhecer, com um único set de dados, alterações genéticas e epi-genéticas neste tipo. de câncer. Sequenciador: 454.")
17
Fapesp/LICR Genoma Humano do Câncer Projeto 454 Venter/LICR # sequências 1.2 milhões mil # sequenciadores 05 MegaBaces # corridas ~15, Custo (US$)* 12 milhões 10 mil * Excluindo o preço dos aparelhos
* 12 milhões 10 mil. * Excluindo o preço dos aparelhos.")
18
Um objetivo a curto prazo

19
Os sequenciadores de nova geração promovem uma mudança no paradigma
Geração de dados deixa de ser o fator limitante Com os bilhões de datapoints gerados em horas, o processamento e análise dos dados tornou-se o maior gargalo das pesquisas biomédicas.

20
Bioinformática Computação Matemática Biologia

21
O que é Bioinformática?

24
Bioinformática - História
1970, Needleman/Wunch, Alinhamento Global. 1972, Margaret Dayhoff, Matrizes de Comparação. 1979, Walter Goad, GenBank. 1981, Smith/Waterman, Alinhamento Local. 1989, NHGRI, Projeto Genoma Humano. 1990, Altschul/Gish/Miller/Myers/Lipman, BLAST. 1994, Eddy/Krogh/Durbin, Hidden Markov Models (HMMs).
.")
25
Bioinformática - Importância
Poucas pessoas adequadamente treinadas em Biologia e Computação. Biologia em larga-escala. Produção de dados em massa gera uma demanda para análises computacionais. Economiza tempo e dinheiro.

26
Bioinformática Desenvolvimento de ferramentas.
Forma de explorar novos dados. Processamento de dados gerados por projetos em larga-escala. Uma nova forma de se fazer ciência dirigida por hipóteses.

27
Bioinformática O Bioinformata O Usuário Manipula a informação.
Desenvolve ferramentas Bancos de dados locais. Local. Mta programação. Habilidades de TI. Recursos da Web. Local ou remoto. nada de programação. pouca habilidade de TI.

28
Cinco websites que todos devem conhecer
NCBI (The National Center for Biotechnology Information; EBI (The European Bioinformatics Institute) The UCSC Genome Browser SwissProt/ExPASy (Swiss Bioinformatics Resource) PDB (The Protein Databank)
 The UCSC Genome Browser. SwissProt/ExPASy (Swiss Bioinformatics Resource) PDB (The Protein Databank)")
29
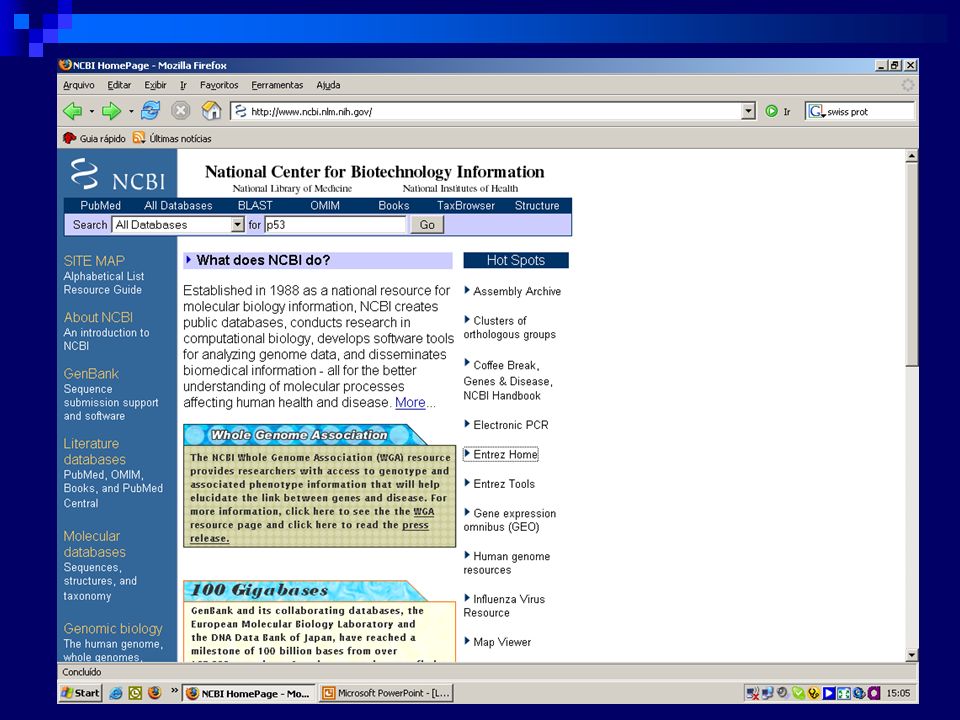
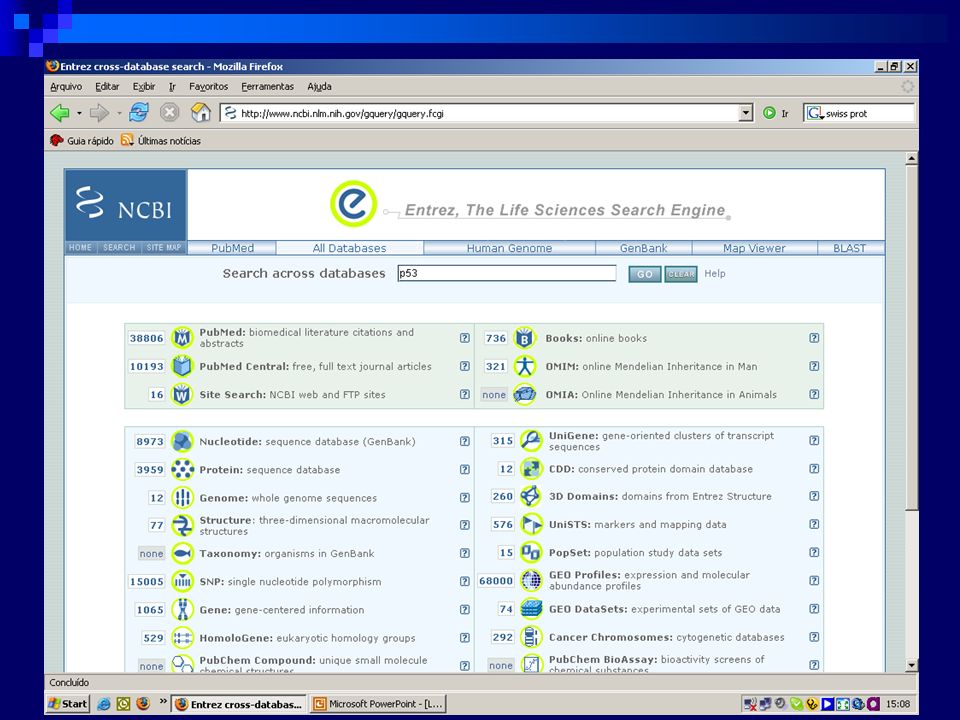
NCBI (http://www.ncbi.nlm.nih.gov/)
Acesso aos bancos de dados via Entrez Medline/OMIM Genbank/Genpept/Structures Servidor de BLAST Todos os tipos de Blast Portal do Genoma Humano Muito, muito mais……..

32
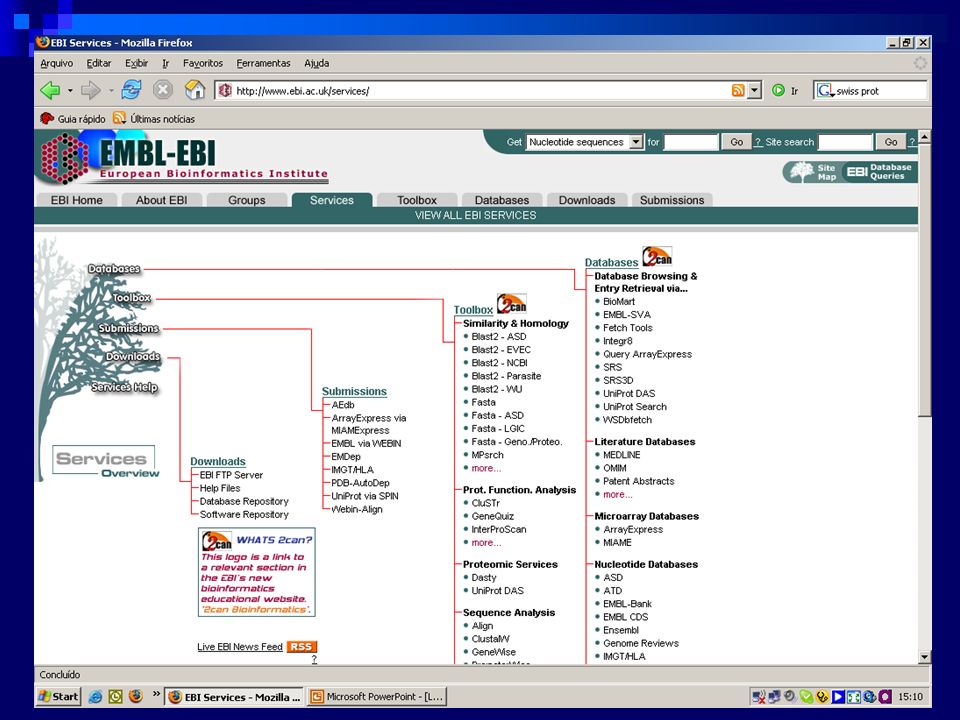
EBI (http://www.ebi.ac.uk/)
Acesso a bancos de dados via SRS EMBL, SwissProt, …… Muitas outras ferramentas ClustalW, DALI, …

34
UCSC Genome Browser (http://genome.ucsc.edu/)
Banco de dados e Browser para genomas de diferentes espécies Humano, camundongo, rato, zebrafish, etc…. Muitas outras ferramentas SNPs, domínios prtéicos, genômica comparativa, etc….

36
SwissProt (http://www.expasy.ch/sprot/)
Checagem manual. O número de entradas errôneas é bastante reduzido. Cross-link extensivo com outros bancos SwissProt é o ‘gold-standard’ em termos de bancos de dados e é o melhor lugar para se começar uma análise se vc procura info para uma ou poucas

38
Protein Data Bank – PDB (http://www.rcsb.org/pdb/)
Armazena a estrutura tri-dimensional para milhares de proteínas Acesso a vários serviços relacionados a biologia estrutural

40
Bancos de Sequência Primários
GenBank (USA) EMBL (Europa) DDBJ (Japão)
 EMBL (Europa) DDBJ (Japão)")
41
Homologia - Ortologia - Paralogia

42
Dois conceitos importantes
Paralogia: O evento que originou às duas sequências é um evento de duplicação gênica! Orthologia: O evento que deu origem às duas sequências é um evento de especiação! FUNÇÕES SIMILARES! FUNÇÕES IDÊNTICAS!

43
Como definir função? Alinhamento de sequências
Motivos (padrões consensuais) Blocos, perfis, etc.... Hidden Markov Models - HMM Since it is difficult to directly predict structure and function, various representations are used to look at different angles. Sequence alignment attempts to align sequences with known proteins. If they match to some degree, there may be similarities in function. Motifs are in essence regular expressions that correspond to known short sequences that have specific binding properties. These can be used to glean more information. Blocks, profiles, templates are probabalistic ways of representing motifs. Hidden Markov models are used to detect relationships between proteins.
 Blocos, perfis, etc.... Hidden Markov Models - HMM. Since it is difficult to directly predict structure and function, various representations are used to look at different angles. Sequence alignment attempts to align sequences with known proteins. If they match to some degree, there may be similarities in function. Motifs are in essence regular expressions that correspond to known short sequences that have specific binding properties. These can be used to glean more information. Blocks, profiles, templates are probabalistic ways of representing motifs. Hidden Markov models are used to detect relationships between proteins.")
44
Similarity Searches on Sequence Databases, EMBnet Course, October 2003

45
Alinhamento 0 1 2 3 4 5 6 7 8 9 10 G A A - G G A T T A G
G A A - G G A T T A G G A T C G G A A G Identidade - MATCH Semelhança / divergência - MISMATCH Lacunas - GAPS Inserção/Deleção - INDELS Um alinhamento basicamente consiste em acrescentar espacos nas duas sequencias, de tal forma que ambas fiquem do mesmo tamanho. O ideal e que o alinhamento evidencie o emparelhamento de trechos das sequencias que sejam similares, enquanto que os espacos aparecam bem mais em trechos nao-similares. Uma lacuna pode ser definida como sendo um conjunto máximo e consecutivo de espaços que ocorrem numa seqüência quando se tenta alinhá-la a outra, As lacunas ajudam a criar alinhamentos que melhor se conformam aos modelos biológicos e se ajustam bem às buscas de padrões. Um Since there are many sequences with unknown structure and function and a small number of sequences of which the structure and function are known, try matching the unknown sequences against known sequences. If there is a close alignment, there might be close similarities to the known structure/function.

46
Alinhamento Qual é o melhor alinhamento ? A – C – G G – A C T
| | | | | A T C G G A T – C T Alinhamento 2: A T C G G A T C T | | | | | | A – C G G – A C T

47
Pontuação Esquema de pontuação match: +2 mismatch: +1 indel: –2
Alinhamento 1: (5 *2) + (1*1) + (4*-2) = – 8 = 3 Alinhamento 2: (6 *2) + (1*1) + (2*-2) = – 4 = 9 Escore final = soma dos escores para cada posição Favorece os matches, penaliza os gaps
 + (1*1) + (4*-2) = – 8 = 3. Alinhamento 2: (6 *2) + (1*1) + (2*-2) = – 4 = 9. Escore final = soma dos escores para cada posição. Favorece os matches, penaliza os gaps.")
48
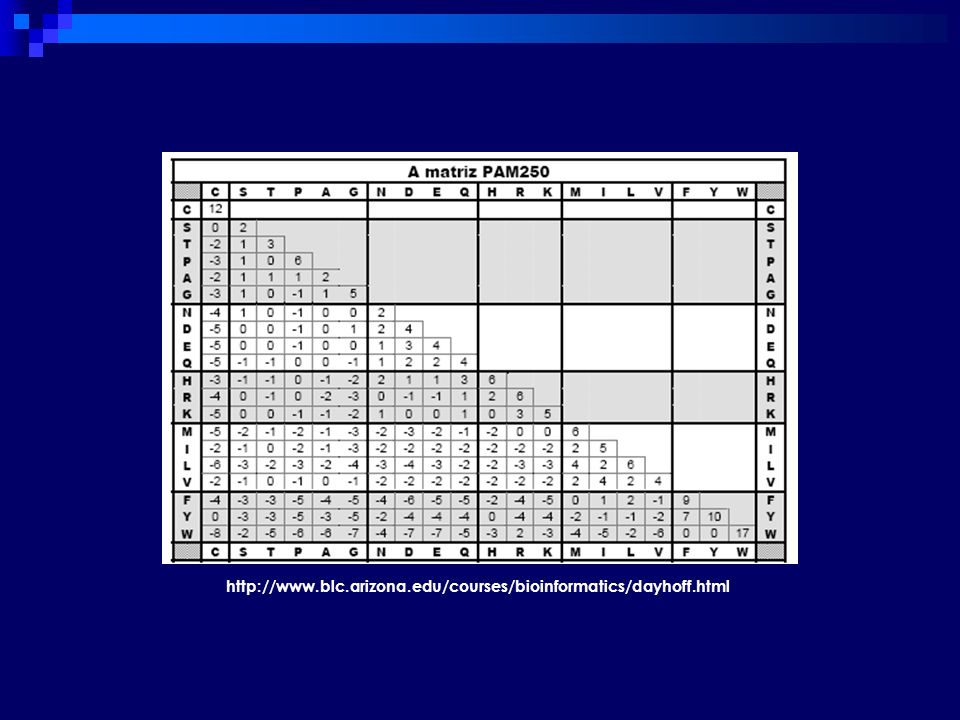
Matriz de Substituição
Tabela de comparação Reflete a probabilidade ou frequência de determinada substituição em sequências biologicamente relacionadas p(A B) = p(B A) Construídas pelo estudo do alinhamento de diversas sequências relacionadas AA ou nucleotídeos São tabelas bidimensionais (i, j) que contêm valores que demonstram a probabilidade de que o aminoácido da posição i sofra mutação para o aminoácido da posição j, quaisquer que sejam as duas seqüências envolvidas. Para alinhamento de proteınas, o metodo de pontuacao simples aplicado ao DNA nao e suficiente. Os aminoacidos possuem propriedades bioquımicas que determinam como eles sao substituıdos durante a evolucao. Por exemplo, existe uma maior probabilidade de que um aminoacido seja substituıdo por um outro de igual tamanho em lugar de um aminoacido maior. Dado que a comparacao de proteınas e feita frequentemente com criterios evolutivos, e necessario um esquema de pontuacao que leve em conta estas probabilidades Se a amostra é grande o suficiente para ser estatisticamente significante, as matrizes devem refletir as verdadeiras possibilidades de mutações que ocorreram ao longo de um certo período de evolução.
 = p(B A) Construídas pelo estudo do alinhamento de diversas sequências relacionadas. AA ou nucleotídeos. São tabelas bidimensionais (i, j) que contêm valores que demonstram a probabilidade de que o aminoácido da posição i sofra mutação para o aminoácido da posição j, quaisquer que sejam as duas seqüências envolvidas. Para alinhamento de proteınas, o metodo de pontuacao simples aplicado ao DNA nao e suficiente. Os aminoacidos possuem propriedades bioquımicas que determinam como eles sao substituıdos durante a evolucao. Por exemplo, existe uma maior probabilidade de que um aminoacido seja substituıdo por um outro de igual tamanho em lugar de um aminoacido maior. Dado que a comparacao de proteınas e feita frequentemente com criterios evolutivos, e necessario um esquema de pontuacao que leve em conta estas probabilidades. Se a amostra é grande o suficiente para ser estatisticamente significante, as matrizes devem refletir as verdadeiras possibilidades de mutações que ocorreram ao longo de um certo período de evolução.")
49
Percent Accepted Mutation (PAM - Dayhoff)
Margaret Dayhoff (1978) Probabilidade de substituição de aa em alinhamentos globais de sequências homólogas Cada matriz reflete as mutações entre sequências que divergiram por determinado período de tempo Mutações aceitas => não afetam negativamente a viabilidade da proteína Primeira matriz 71 grupos de proteínas, 85% de similaridade 1572 substitutições de aminoácidos Expansão do número de proteínas => 1991 database
 Probabilidade de substituição de aa em alinhamentos globais de sequências homólogas. Cada matriz reflete as mutações entre sequências que divergiram por determinado período de tempo. Mutações aceitas => não afetam negativamente a viabilidade da proteína. Primeira matriz. 71 grupos de proteínas, 85% de similaridade substitutições de aminoácidos. Expansão do número de proteínas => 1991 database.")
50
Matrizes PAM Premissa => cada mutação é independente das mutações anteriores Consequência => as substituições observadas em curtos períodos podem ser extrapoladas para longos períodos PAM 1 => sequências com 1% ou menos de divergência =>1 mutação aceita a cada 100 aminoácidos PAM N mutações = (PAM 1)N PAM 250 => 250 mutações por 100 aa => 250% mutações em 2500 milhões anos PAM 250: 20% similar - PAM 120: 40% - PAM 80: 50% - PAM 60: 60%
N. PAM 250 => 250 mutações por 100 aa => 250% mutações em 2500 milhões anos. PAM 250: 20% similar - PAM 120: 40% - PAM 80: 50% - PAM 60: 60%")
52
Blocks Substitution Matrix (BLOSUM)
Kenikoff & Henikoff (1992) Frequência de substituição de aa em um conjunto de ~2000 padrões (blocos) Maior número de sequências consideradas => mais de 500 famílias Alinhamentos locais de sequências relacionadas e não geradas a partir de extrapolações BLOSUM 62 é o padrão para BLAST 2.0 => sequências moderadamente distantes ou mais próximas Sequências Consenso 60% idênticas: BLOSUM 60 80% idênticas : BLOSUM 80
 Frequência de substituição de aa em um conjunto de ~2000 padrões (blocos) Maior número de sequências consideradas => mais de 500 famílias. Alinhamentos locais de sequências relacionadas e não geradas a partir de extrapolações. BLOSUM 62 é o padrão para BLAST 2.0 => sequências moderadamente distantes ou mais próximas. Sequências Consenso. 60% idênticas: BLOSUM % idênticas : BLOSUM 80.")
53
G A V C T K I G V V C Y R E 6+0+4+9+(-2)+2+(-3)= 16
G A V C T K I G V V C Y R E (-2)+2+(-3)= 16
+2+(-3)= 16.")
54
Relação ente BLOSUM e PAM
PAM => origens evolutivas de proteínas BLOSUM => domínios conservados

55
Global vs. Local Global Alinhamento de toda a sequência utilizado o maior número de caracteres possíveis Sequências similares e de tamanho aproximado Local Segmentos com o maior número de identidades Regiões alinhadas e não alinhadas (≠ mismatch) Sequências similares em algumas regiões, que diferem em tamanho ou que compartilham domínios conservados
 Sequências similares em algumas regiões, que diferem em tamanho ou que compartilham domínios conservados.")
56
Aplicações Global Deduzir histórias evolutivas entre membros da mesma família Estabelecer a existência de um ancestral comum (homologia) Local Inferir funções biológicas Identificar regiões conservadas e de alta similaridade (sítio ativo, domínios) entre outras pouco conservadas Reconstruir sequências de DNA a partir de seus fragmentos Comparar sequências de mRNA (sem íntrons) à sequência genômica
 entre outras pouco conservadas. Reconstruir sequências de DNA a partir de seus fragmentos. Comparar sequências de mRNA (sem íntrons) à sequência genômica.")
57
Métodos de Análise Diagramas - DOT PLOT
Algoritmo de Programação Dinâmica Algoritmos Heurísticos - Word-Based ou K-tuples

58
Dot Plot

59
Inserções & Deleções

60
Repetições & Inversões

61
Programas Disponíveis
Dotter ( COMPARE & DOTPLOT (Genetics Computer Group) PLALIGN ( Web browser (
 PLALIGN ( Web browser (")
62
Programação Dinâmica Needleman & Wunsch (1970)
Compara cada par de caracteres nas duas sequências Posiciona os gaps de forma a obter o maior número de alinhamentos idênticos ou similares Gera uma matriz de números que representa todos os possíveis alinhamentos de acordo com um sistema de escore Alinhamento ótimo => maior escore

63
Limitações Computacionalmente lento
Número de alinhamentos cresce exponencialmente com a média dos comprimentos das sequências (n) Número de cálculos => proporcional a n2 ou n3 Memória => capacidade da ordem de n2
 Número de cálculos => proporcional a n2 ou n3. Memória => capacidade da ordem de n2.")
64
Needleman-Wunsch As sequências abcdefghajklm abbdhijk
São alinhadas e scores são dados a b c d e f g h a j k l m | | | | | | a b b d h i j k match mismatch gap_open gap_extend Score total de = 13.

65
Needleman-Wunsch O alinhamento de maior score entre as duas
sequências é considerado o mais provável.

66
Overall %id = 43.15 Overall %similarity = 60.27
Needleman-Wunsch Saída típica: Global: HBA_HUMAN vs HBB_HUMAN Score: HBA_HUMAN VLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFP 44 |:| :|: | | |||| : | | ||| |: : :| |: :| HBB_HUMAN VHLTPEEKSAVTALWGKV..NVDEVGGEALGRLLVVYPWTQRFFE 43 HBA_HUMAN HF.DLS.....HGSAQVKGHGKKVADALTNAVAHVDDMPNALSAL 83 | ||| |: :|| ||||| | :: :||:|:: : | HBB_HUMAN SFGDLSTPDAVMGNPKVKAHGKKVLGAFSDGLAHLDNLKGTFATL 88 HBA_HUMAN SDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKF 128 |:|| || ||| ||:|| : |: || | |||| | |: | HBB_HUMAN SELHCDKLHVDPENFRLLGNVLVCVLAHHFGKEFTPPVQAAYQKV 133 HBA_HUMAN LASVSTVLTSKYR :| |: | || HBB_HUMAN VAGVANALAHKYH %id = %similarity = 63.31 Overall %id = Overall %similarity = 60.27

67
Razões para se usar um banco de sequências
Eu acabei de obter uma sequência. O que é sabido à respeito desta sequência? Ela é única? Eu tenho uma sequência única. Ela tem similaridade com alguma outra sequência de função conhecida? Eu encontrei uma nova proteína em um determinado organismo. Existe um ortólogo conhecido? Eu decidi trabalhar com um gene novo. Eu não tenho como obter um clone contendo a sequência deste gene. Eu preciso da sequência do cDNA para fazer uma PCR.

68
O que envolve uma busca ? Algoritmos de busca (BLAST, FASTA)
Matrizes de comparação (PAM vs. BLOSUM) Banco de dados (nr, dbEST) Parâmetros de busca (filtros on/off, threshold, etc…)
 Banco de dados (nr, dbEST) Parâmetros de busca (filtros on/off, threshold, etc…)")
69
Basic Local Alignment Search Tool
Método heurístico => método empírico, que utiliza a fórmula ´tentativa e erro´ para encontrar as soluções Significado estatístico => determina se um alinhamento ocorre aleatoriamente ou não Vantagem => pelo menos 50 vezes mais rápido que os algoritmos de programação dinâmica e mais apropriados para busca em bancos de dados Desvantagem => não garante uma solução com um alinhamento ótimo como os algoritmos de programação dinâmica o método mais utilizado para realizar buscas de seqüências similares em bancos de dados de seqüências 2) Heurístico – conjunto de regras e métodos que conduzem à descoberta, à invenção e à resolução de problemas; metodologia ou algoritmo usada para resolver problemas por métodos que, embora não rigorosos, geralmente refletem o conhecimento humano e permitem obter uma solução satisfatória. Tenta privilegiar a eficiência computacional ao mesmo tempo em que otimiza uma medida de similaridade específica The BLAST algorithm is a heuristic program, which means that it relies on some smart shortcuts to perform the search faster. BLAST performs "local" alignments. Most proteins are modular in nature, with functional domains often being repeated within the same protein as well as across different proteins from different species. The BLAST algorithm is tuned to find these domains or shorter stretches of sequence similarity. The local alignment approach also means that a mRNA can be aligned with a piece of genomic DNA, as is frequently required in genome assembly and analysis. If instead BLAST started out by attempting to align two sequences over their entire lengths (known as a global alignment), fewer similarities would be detected, especially with respect to domains and motifs 3) Similarity is not homology, things may be % similar, but they are either homologous or not. Local aligns active sites on proteins, important since most proteins are modular in nature. A global alignment does not take this into account and similarities may be missed. Statistical theory very important as it tells us whether or not an alignment occurred just by chance.
 Heurístico – conjunto de regras e métodos que conduzem à descoberta, à invenção e à resolução de problemas; metodologia ou algoritmo usada para resolver problemas por métodos que, embora não rigorosos, geralmente refletem o conhecimento humano e permitem obter uma solução satisfatória. Tenta privilegiar a eficiência computacional ao mesmo tempo em que otimiza uma medida de similaridade específica. The BLAST algorithm is a heuristic program, which means that it relies on some smart shortcuts to perform the search faster. BLAST performs local alignments. Most proteins are modular in nature, with functional domains often being repeated within the same protein as well as across different proteins from different species. The BLAST algorithm is tuned to find these domains or shorter stretches of sequence similarity. The local alignment approach also means that a mRNA can be aligned with a piece of genomic DNA, as is frequently required in genome assembly and analysis. If instead BLAST started out by attempting to align two sequences over their entire lengths (known as a global alignment), fewer similarities would be detected, especially with respect to domains and motifs. 3) Similarity is not homology, things may be % similar, but they are either homologous or not. Local aligns active sites on proteins, important since most proteins are modular in nature. A global alignment does not take this into account and similarities may be missed. Statistical theory very important as it tells us whether or not an alignment occurred just by chance.")
70
Aplicações Identificar sequências ortólogas e parálogas
Descobrir novos genes ou proteínas Descobrir variantes de genes e proteínas Investigar Expressed Sequence Tags - ESTs Explorar a estrutura e função de proteínas

71
BLAST WEB Pages BLAST (NCBI – National Center for Biotechnology Information): BLAST2 (Swiss EMBnet server - European Molecular Biology network??): WU-BLAST (Washington University): The way most people use BLAST is to input a nucleotide or protein sequence as a query against all (or a subset of) the public sequence databases, pasting the sequence into the textbox on one of the BLAST Web pages. This sends the query over the Internet, the search is performed on the NCBI databases and servers, and the results are posted back to the person's browser in the chosen display format Os dois últimos são BLAST modificados para incorporar gaps na etapa de alongamento das palavras
: WU-BLAST (Washington University): The way most people use BLAST is to input a nucleotide or protein sequence as a query against all (or a subset of) the public sequence databases, pasting the sequence into the textbox on one of the BLAST Web pages. This sends the query over the Internet, the search is performed on the NCBI databases and servers, and the results are posted back to the person s browser in the chosen display format. Os dois últimos são BLAST modificados para incorporar gaps na etapa de alongamento das palavras.")
72
O BLAST oferecido pelo NCBI é, na verdade, uma família de serviços, onde o usuário possui diversas opções, dependendo da seqüência de entrada, se ela é constituída de nucleotídeos ou aminoácidos, se o banco de dados alvo é de nucleotídeos, aminoácidos ou está restrito a um tipo de organismo, além dos parâmetros relacionados ao algoritmo de busca.

73
Blast é Heurístico 1) Tabela de busca com todas as ´palavras´ (words) de comprimento W (3 aa ou11 nucleot.) mais as palavras vizinhas semelhantes, que aparecem pelo menos T vezes na sequência query. 2) Busca de sementes (hits, hot spots) na sequência do banco de dados que alinhem com as palavras previamente estabelecidas. 3) Extensão das sementes em ambas as direções, produzindo alinhamentos locais máximos (HSP - high scoring pair) com ou sem lacunas, de acordo com os parâmetros estabelecidos. 4) Registro da informação em um arquivo SeqAlign (ASN.1). 5) A informação é utilizada para buscar sequências similares. Os resultados podem ser reformatados sem a necessidade de refazer a busca. 2) Ou seja, as duas sequências possuem o mesmo aminoácido na posição correspondente 3) inicialmente era realizada sem considerar gaps atualmente, as extensões são feitas com gaps
 Tabela de busca com todas as ´palavras´ (words) de comprimento W (3 aa ou11 nucleot.) mais as palavras vizinhas semelhantes, que aparecem pelo menos T vezes na sequência query. 2) Busca de sementes (hits, hot spots) na sequência do banco de dados que alinhem com as palavras previamente estabelecidas. 3) Extensão das sementes em ambas as direções, produzindo alinhamentos locais máximos (HSP - high scoring pair) com ou sem lacunas, de acordo com os parâmetros estabelecidos. 4) Registro da informação em um arquivo SeqAlign (ASN.1). 5) A informação é utilizada para buscar sequências similares. Os resultados podem ser reformatados sem a necessidade de refazer a busca. 2) Ou seja, as duas sequências possuem o mesmo aminoácido na posição correspondente. 3) inicialmente era realizada sem considerar gaps atualmente, as extensões são feitas com gaps.")
74
Sensibilidade vs. Seletividade
Habilidade de encontrar a maior parte dos membros relacionados à família da sequência query Seletividade Habilidade de não identificar sequências de outras famílias como falso-positivos “Grau de cobertura dos membros da família dado um nível de falso-positivos” Once BLAST has found a similar sequence to the query in the database, it is helpful to have some idea of whether the alignment is "good" and whether it portrays a possible biological relationship, or whether the similarity observed is attributable to chance alone. BLAST uses statistical theory to produce a bit score and expect value (E-value) for each alignment pair (query to hit). Although a statistician might consider this to be significant, it still may not represent a biologically meaningful result, and analysis of the alignments (see below) is required to determine "biological" significance
 for each alignment pair (query to hit). Although a statistician might consider this to be significant, it still may not represent a biologically meaningful result, and analysis of the alignments (see below) is required to determine biological significance.")
75
Escores e Estatística Bit Score
Indica quão bom é o alinhamento. Quanto maior o escore, melhor o alinhamento Considera o número de resíduos idênticos ou similares e a quantidade de gaps Influenciado pela Matriz de Substituição (padrão: BLOSUM 62 ) Exceção: blastn and MegaBLAST Normalização: bit scores de diferentes alinhamentos podem ser comparados Once BLAST has found a similar sequence to the query in the database, it is helpful to have some idea of whether the alignment is "good" and whether it portrays a possible biological relationship, or whether the similarity observed is attributable to chance alone. BLAST uses statistical theory to produce a bit score and expect value (E-value) for each alignment pair (query to hit). Although a statistician might consider this to be significant, it still may not represent a biologically meaningful result, and analysis of the alignments (see below) is required to determine "biological" significance
 Exceção: blastn and MegaBLAST. Normalização: bit scores de diferentes alinhamentos podem ser comparados. Once BLAST has found a similar sequence to the query in the database, it is helpful to have some idea of whether the alignment is good and whether it portrays a possible biological relationship, or whether the similarity observed is attributable to chance alone. BLAST uses statistical theory to produce a bit score and expect value (E-value) for each alignment pair (query to hit). Although a statistician might consider this to be significant, it still may not represent a biologically meaningful result, and analysis of the alignments (see below) is required to determine biological significance.")
76
Escores e Estatística E-value Significado estatístico do alinhamento
Quanto menor o escore, mais significativo é o alinhamento E-value = Significa que existem 5 chances em 100 (1 em 20) da similaridade entre as sequências ocorrer aleatoriamente Influenciado pelo tamanho do banco de dados e o sistema de escore utilizado Once BLAST has found a similar sequence to the query in the database, it is helpful to have some idea of whether the alignment is "good" and whether it portrays a possible biological relationship, or whether the similarity observed is attributable to chance alone. BLAST uses statistical theory to produce a bit score and expect value (E-value) for each alignment pair (query to hit). Although a statistician might consider this to be significant, it still may not represent a biologically meaningful result, and analysis of the alignments (see below) is required to determine "biological" significance
 da similaridade entre as sequências ocorrer aleatoriamente. Influenciado pelo tamanho do banco de dados e o sistema de escore utilizado. Once BLAST has found a similar sequence to the query in the database, it is helpful to have some idea of whether the alignment is good and whether it portrays a possible biological relationship, or whether the similarity observed is attributable to chance alone. BLAST uses statistical theory to produce a bit score and expect value (E-value) for each alignment pair (query to hit). Although a statistician might consider this to be significant, it still may not represent a biologically meaningful result, and analysis of the alignments (see below) is required to determine biological significance.")
77
Etapas de Busca 1) Selecionar a sequência (query)
2) Selecionar o banco de dados 3) Selecionar o programa 4) Definir os parâmetros
 Selecionar o banco de dados. 3) Selecionar o programa. 4) Definir os parâmetros.")
78
Passo 1: Escolha da sequência
Natureza Tamanho Formatos : Identificadores (ID), FASTA (>seq name), sequências puras (txt?) ID : códigos para acesso aos bancos de dados mantidos pelo NCBI Txt: intercaladas ou não por caracteres brancos ou numéricos
, FASTA (>seq name), sequências puras (txt ) ID : códigos para acesso aos bancos de dados mantidos pelo NCBI. Txt: intercaladas ou não por caracteres brancos ou numéricos.")
81
Passo 2: Seleção do Banco de Dados
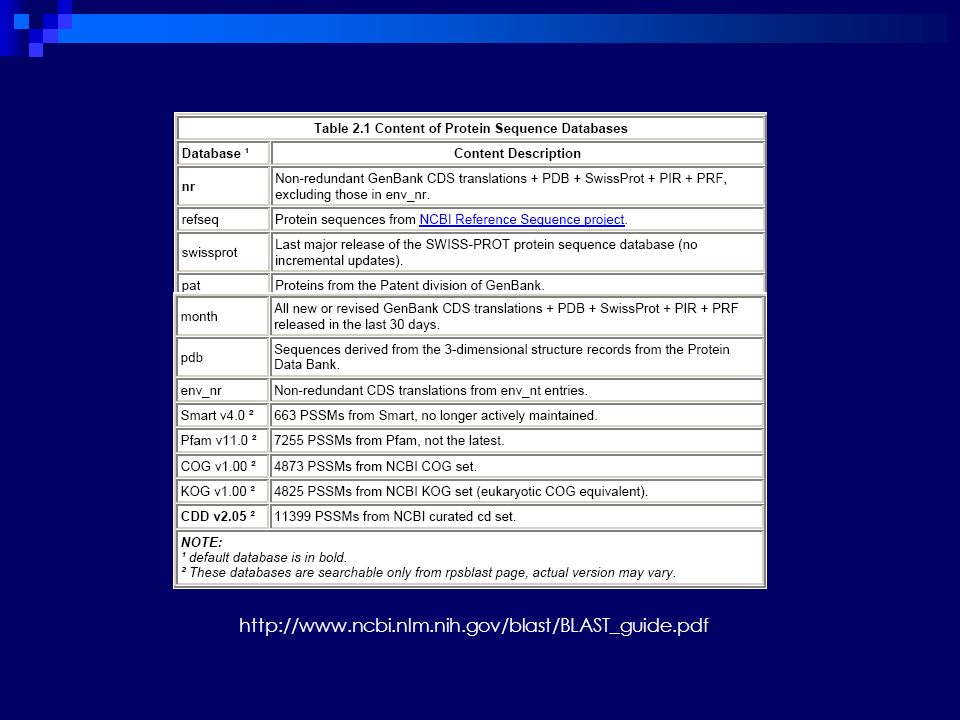
Proteínas GenBank, PDB, SWISSPROT, PIR, REPBASE68 e BDGP Nucleotídeos GenBank, EMBL, DDBJ, PDB, REPBASE, BDGP, EST69, STS70, vetores, sequências de mitocôndrias, GSS71, sequências HTGS72 Conteúdo não-redundância, periodicidade de atualização organismos ou espécies sequências patenteadas interesse imunológico elementos repetitivos, etc. TIPO & CONTEUDO ou organização

84
Passo 3: Seleção do Programa
Natureza da sequência Finalidade da busca Banco de dados Levar em conta 3 parametros

85
DNA codifica 6 proteínas potenciais
5’ CAT CAA 5’ ATC AAC 5’ TCA ACT 5’ CATCAACTACAACTCCAAAGACACCCTTACACATCAACAAACCTACCCAC 3’ 3’ GTAGTTGATGTTGAGGTTTCTGTGGGAATGTGTAGTTGTTTGGATGGGTG 5’ 5’ GTG GGT 5’ TGG GTA 5’ GGG TAG

86
Tipos de Programas programa entrada banco de dados 1 blastn DNA DNA
blastp protein protein 6 blastx DNA protein tblastn protein DNA 36 tblastx DNA DNA

87
Passo 4: Seleção dos parâmetros
Tamanho da palavra (w-er) Filtros E value Matriz de substituição & penalidades para gap BLAST oferece valores default para uma série de parâmetros utilizados pelo algoritmo de busca, entretanto todos são configuráveis pelo usuário 1) indica o nível de significância a partir do qual os resultados serão incluídos 3) Dependendo da distância evolucionária desejada uma matriz pode ser mais adequada que outra. Para a matriz PAM, maior n corresponde a maior distância evolucionária, enquanto que para matrizes BLOSUM mantém-se a relação inversa Sensibilidade e velocidade => W, T e X Seletividade => cutoff score
 Filtros. E value. Matriz de substituição & penalidades para gap. BLAST oferece valores default para uma série de parâmetros utilizados pelo algoritmo de busca, entretanto todos são configuráveis pelo usuário. 1) indica o nível de significância a partir do qual os resultados serão incluídos. 3) Dependendo da distância evolucionária desejada uma matriz pode ser mais adequada que outra. Para a matriz PAM, maior n corresponde a maior distância evolucionária, enquanto que para matrizes BLOSUM mantém-se a relação inversa. Sensibilidade e velocidade => W, T e X. Seletividade => cutoff score.")
88
Entrez Filter Expect organism Word size Scoring matrix

89
Filtro

90
Report Header Tipo de programa (BLASTP), versão (2.2.1) e data da versão Artigo que descreve o BLAST, request ID (issued by QBLAST), a definição da sequência e resumo do banco de dados Taxonomy reports: mostra o resultado deste BLAST na base de informação do banco de dados Taxonomy
, a definição da sequência e resumo do banco de dados. Taxonomy reports: mostra o resultado deste BLAST na base de informação do banco de dados Taxonomy.")
91
Taxonomy Report

92
Graphical Overview query database hits
In this case, there are three high-scoring database matches that align to most of the query sequence. Mousing over the bars displays the definition line for that sequence to be shown in the window above the graphic Quanto mais próximas da query, mais semelhantes Barras em rosa: lower-scoring matches que alinham em 2 regiões (resíduos 3-60 e ) Segmento rachurado: as duas regiões de similaridade estão na mesma proteína mas esta região não alinha Outras barras: lower-scoring matches
 Segmento rachurado: as duas regiões de similaridade estão na mesma proteína mas esta região não alinha. Outras barras: lower-scoring matches.")
93
One-line Descriptions
For the first hit in the list, the gi number is , the database designation is sp (for SWISS-PROT), the Accession number is P26374, the locus name is RAE2_HUMAN, the definition line is Rab proteins, the score is 1216, and the E-value is 0.0. Note that the first 17 hits have very low E-values (much less than 1) and are either RAB proteins or GDP dissociation inhibitors. The other database matches have much higher E-values, 0.5 and above, which means that these sequences may have been matched by chance alone. This usually includes information on the organism from which the sequence was derived, the type of sequence (e.g., mRNA or DNA), and some information about function or phenotype. The definition line is often truncated in the one-line descriptions to keep the display compact bitsHigher scoring hits are found at the top of the list; E-value which provides an estimate of statistical significance (a) gi number, designação do banco de dados, número de acesso e o nome do locus para as sequências encontradas, separados por barras verticais (b) Definição da sequência (c) Escore de alinhamento ( bits) (d) E-value
, the Accession number is P26374, the locus name is RAE2_HUMAN, the definition line is Rab proteins, the score is 1216, and the E-value is 0.0. Note that the first 17 hits have very low E-values (much less than 1) and are either RAB proteins or GDP dissociation inhibitors. The other database matches have much higher E-values, 0.5 and above, which means that these sequences may have been matched by chance alone. This usually includes information on the organism from which the sequence was derived, the type of sequence (e.g., mRNA or DNA), and some information about function or phenotype. The definition line is often truncated in the one-line descriptions to keep the display compact. bitsHigher scoring hits are found at the top of the list; E-value which provides an estimate of statistical significance. (a) gi number, designação do banco de dados, número de acesso e o nome do locus para as sequências encontradas, separados por barras verticais. (b) Definição da sequência. (c) Escore de alinhamento ( bits) (d) E-value.")
94
Pairwise Sequence Alignment

96
Famílias de Elementos Repetitivos
Alu L1 L2 Tais sequências podem gerar alinhamentos espúrios.

97
Alu Constitutes about 5% of the human genome.
Short interspersed repeats. Found in primate genomes. ALU elements often found in 3’ regions or introns.

98
Blast usando uma sequência de Alu

100
Como identificar e remover elementos repetitivos
Filter para elementos repetitivos no servidor de Blast do NCBI Repeat Masker:

101
Nair & Rost, 2002

102
Way out! psi-Blast pattern (phi-Blast) Hidden Markov Models (HMMs)
 Hidden Markov Models (HMMs)")
103
Position Specific Interactive (PSI)-BLAST
Detecta proteínas fracamente relacionadas ou novos membros de uma família protéica (mais sensível) Utilizado quando o BLAST padrão falha em encontrar hits significativos ou retorna hits com decrições do tipo "hypothetical protein" ou "similar to... " Busca iterativa => comparam-se as sequências de alto escore com a sequência de busca para determinar quais delas são altamente conservadas Sequências resultantes => construção de um modelo de escore específico por posição (consenso) => Position-Specific Scoring Matrix (PSSM ou profile)
 Utilizado quando o BLAST padrão falha em encontrar hits significativos ou retorna hits com decrições do tipo hypothetical protein ou similar to... Busca iterativa => comparam-se as sequências de alto escore com a sequência de busca para determinar quais delas são altamente conservadas. Sequências resultantes => construção de um modelo de escore específico por posição (consenso) => Position-Specific Scoring Matrix (PSSM ou profile)")
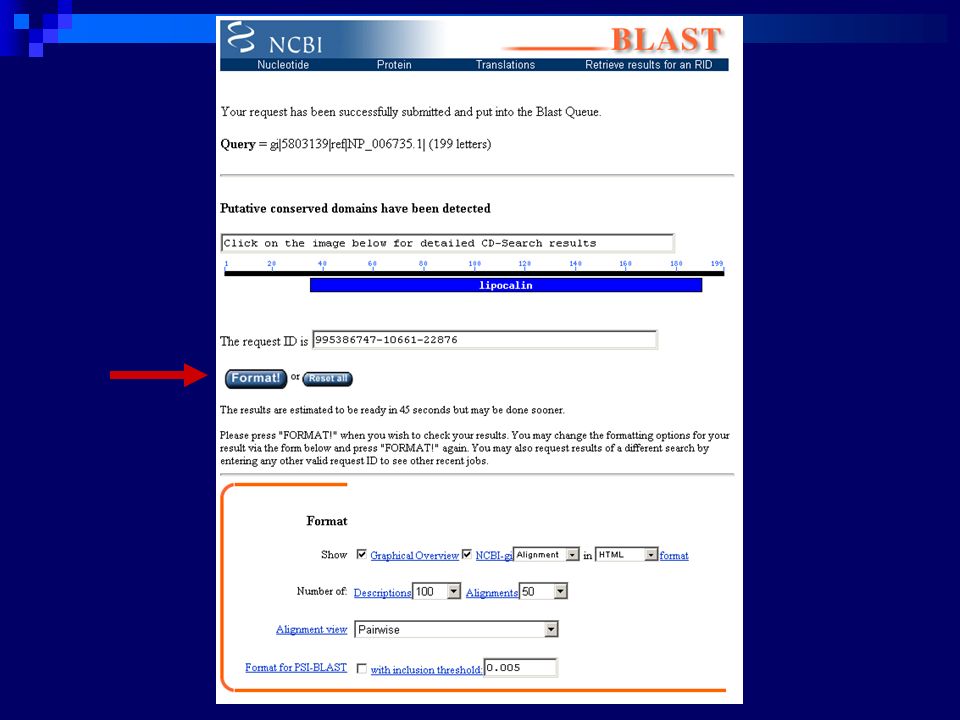
104
PSI-BLAST - Algoritmo Busca com BLASTp normal
Construção de um consenso a partir das regiões alinhadas com E values menores que o limite estabelecido (padrão = 0.005) Utilizando este consenso, procede a uma nova pesquisa sobre a base de dados Quaisquer novos hits abaixo do limite são incluídos em um novo PSSM Fim do processo (convergência) : nenhuma nova sequência é adicionada ao consenso em iterações subsequentes A cada interação o usuário pode remover algumas das sequências do conjunto de respostas da interação anterior, bem como salvar o profile corrente You can add database hits that fall outside the inclusion threshold to your PSSM for the next round by checking the box next to the hit Already selected hits can also be removed from the selection by uncheck the checkbox
 Utilizando este consenso, procede a uma nova pesquisa sobre a base de dados. Quaisquer novos hits abaixo do limite são incluídos em um novo PSSM. Fim do processo (convergência) : nenhuma nova sequência é adicionada ao consenso em iterações subsequentes. A cada interação o usuário pode remover algumas das sequências do conjunto de respostas da interação anterior, bem como salvar o profile corrente. You can add database hits that fall outside the inclusion threshold to your PSSM for the next round by checking the box next to the hit. Already selected hits can also be removed from the selection by uncheck the checkbox.")
105
Pattern-Hit Initiated (PHI)-BLAST
Busca proteínas que contém padrão especificado pelo usuário E é similar à sequência query em relação in the vicinity ao padrão Reduz o número de hits que contém o padrão no banco de dados mas pode também apresentar nenhuma homologia ao query Exemplo de sequência query e um padrão no formato ProSite: >gi| |ref|NP_ | Human cAMP-dependent protein kinase MSHIQIPPGLTELLQGYTVEVLRQQPPDLVEFAVEYFTRLREARAPASVLPAATPRQSLGHPPPEPGPDR VADAKGDSESEEDEDLEVPVPSRFNRRVSVCAETYNPDEEEEDTDPRVIHPKTDEQRCRLQEACKDILLF KNLDQEQLSQVLDAMFERIVKADEHVIDQGDDGDNFYVIERGTYDILVTKDNQTRSVGQYDNRGSFGELA LMYNTPRAATIVATSEGSLWGLDRVTFRRIIVKNNAKKRKMFESFIESVPLLKSLEVSERMKIVDVIGEK IYKDGERIITQGEKADSFYIIESGEVSILIRSRTKSNKDGGNQEVEIARCHKGQYFGELALVTNKPRAAS AYAVGDVKCLVMDVQAFERLLGPCMDIMKRNISHYEEQLVKMFGSSVDLGNLGQ Padrão encontrado: [LIVMF]-G-E-x-[GAS]-[LIVM]-x(5,11)-R-[STAQ]-A-x-[LIVMA]-x-[STACV]
-R-[STAQ]-A-x-[LIVMA]-x-[STACV]")
106
Hidden Markov Models An approach based on statistical sampling theory
Previously used with success for natural language processing Model sequence as a Markov model that is not known (hidden) Observed sequence is a noisy representation of the hidden “true” model
 Observed sequence is a noisy representation of the hidden true model.")
107
A HMM for a DNA sequence C C T A T (prob 0.8) or A (prob 0.2)
G (prob 0.1) or C (prob 0.9) Each horizontal path is the transition path of the states, and the outwardly visible values are noisy versions of the “True” state values. The true sequence is not known, but observing the noisy values gives clues to what the true value may be. A (prob 0.7) or T (prob 0.3) G (prob 0.1) or C (prob 0.9)
 or C (prob 0.9) Each horizontal path is the transition path of the states, and the outwardly visible values are noisy versions of the True state values. The true sequence is not known, but observing the noisy values gives clues to what the true value may be. A (prob 0.7) or T (prob 0.3) G (prob 0.1) or C (prob 0.9)")
108
Idea of HMM Since multiple alignment of k sequences take O(Nk) time, instead estimate a statistical model of the sequences Align the multiple sequences to this model This is equivalent to aligning the sequences to one another

109
Protein Family Classification
Pfam large collection of multiple sequence alignments and hidden Markov models covers many common protein domains and families Over 73% of all known protein sequences have at least one match 5,193 different protein families

110
Pfam Initial multiple alignment of seeds using a program such as Clustal Alignment hand scrutinized and adjusted

111
Pfam Links to the Pfam software: View some examples:
View some examples:

112
Locating ORFs Simplest method of predicting coding regions is to search for open reading frames (ORFs) open reading frames begin with a start (AUG) codon, and ends with one of three stop codons Six total reading frames
 codon, and ends with one of three stop codons. Six total reading frames.")
113
Locating ORFs Prokaryotes: DNA sequences coding for proteins generally transcribed into mRNA which is translated into protein with very little modification Locating an open reading frame from a start codon to a stop codon can give a strong suggestion into protein coding regions Longer ORFs are more likely to predict protein-coding regions than shorter ORFs.

114
Locating ORFs Eukaryotes: mRNA undergoes processing to remove introns before the protein is translated ORF corresponding to a gene may contain regions with stop codons found within intronic regions Posttranscriptional modification makes gene prediction more difficult

115
Filogenia Problema de determinação de árvores filogenéticas
Encontrar a árvore que melhor descreve a relação entre um conjunto de objetos (espécies ou táxons) Cenoura Baleia Chimpanzé Humano
 Cenoura. Baleia. Chimpanzé. Humano.")
116
Filogenia Táxons e Complexidade 3 árvores possíveis para 4 táxons

117
Filogenia Táxons e Explosão Combinatorial

118
Métodos para reconstrução filogenética
03 métodos principais: : Parsimônia Métodos baseados em distância Verossimilhança máxima

119
Parsimônia Dá preferência à topologia que requer o menor número de mudanças .

120
Filogenia Métodos de Distância
A distância evolutiva é calculada para todos os pares de táxons Matriz de Distâncias A árvore filogenética é construída considerando a relação entre esses valores de distâncias

121
O método de evolução mínima
Para todas as topologias possíveis : Calcula o comprimento de todos os ramos, S Mantém a árvore com menos S. Problema: computacionalmente intenso. Não é usado com mais de 25 sequências.

122
Filogenia Métodos com Critério de Ótimo Máxima Verossimilhança
Determina-se a probabilidade de um modelo evolutivo gerar um certo dado Considera todos os sítios e todas as possibilidades de mutações em todos os nós internos da árvore proposta Multiplica-se a probabilidade de cada sítio Probabilidade da árvore Pode ser utilizado para análises de características e de valores Mais consistente e com estimativas com menor variância Não é simples e intuitivo Computacionalmente intenso

123
Bootstrap procedure O suporte para cada ramo interno é expresso em
termos the % de réplicas.

124
"bootstrapped” tree

125
Bootstrap Ramos internos suportados por ≥ 90% das réplicas são considerados estatisticamente significativos. O procedimento de bootstrap não define se um programa é bom. Uma árvore errada pode ter 100% de suporte de bootstrap em seus ramos internos.

126
Tempo de processamento para vários programas
distance < parsimony ~ PHYML << Bayesian < classical ML NJ DNAPARS PHYML MrBayes fastDNAml,PAUP

127
Recursos de Web para filogenia
Compilações Uma lista de web sites Uma lista grande de programas phylip/software.html

128
Recursos de Web para filogenia
Editor de alinhamento SEAVIEW : para windows e unix Programas para filogenia molecular PHYLIP : PAUP : PHYLO_WIN : MrBayes : PHYML :

129
Recursos de Web para filogenia
Desenho de árvores NJPLOT (para todas as plataformas) Aulas de filogenia
 Aulas de filogenia")
131
Ontologia Fornecer um vocabulário estruturado
e controlado para representar o conhecimento biológico nos bancos de dados.for the

132
Gene Onthology (GO) Biological Process Molecular Function
Objetivo dentro da célula, tecido… Molecular Function Função básica ou tarefa Cellular Component Compartimento ou complexo GO is the designation of a project as well as the product of the project. Starting with the cellular level, we are not distinguishing cell types, organs, etc. Gene Ontology is a collaboration between the fly (FlyBase), mouse (MGD) genome databases, and yeast (SGD). All three groups had started independent projects to produce controlled vocabularies for the biology of their organisms. You will all be familiar with hierarchical system to classify enzymes (EC) or functions (YPD, SwissPROT, MIPS, …). We have divided our project into the creation of three ontologies. These are not necessarily hierarchical rather they can be a network of associations -- a directed acyclic graph (DAG). Process: cell cycle, nutrient transport, behavior, Function: alcohol dehydrogenase, Cellular Location: organelle, protein complex, subcellular compartment
, mouse (MGD) genome databases, and yeast (SGD). All three groups had started independent projects to produce controlled vocabularies for the biology of their organisms. You will all be familiar with hierarchical system to classify enzymes (EC) or functions (YPD, SwissPROT, MIPS, …). We have divided our project into the creation of three ontologies. These are not necessarily hierarchical rather they can be a network of associations -- a directed acyclic graph (DAG). Process: cell cycle, nutrient transport, behavior, Function: alcohol dehydrogenase, Cellular Location: organelle, protein complex, subcellular compartment.")
134
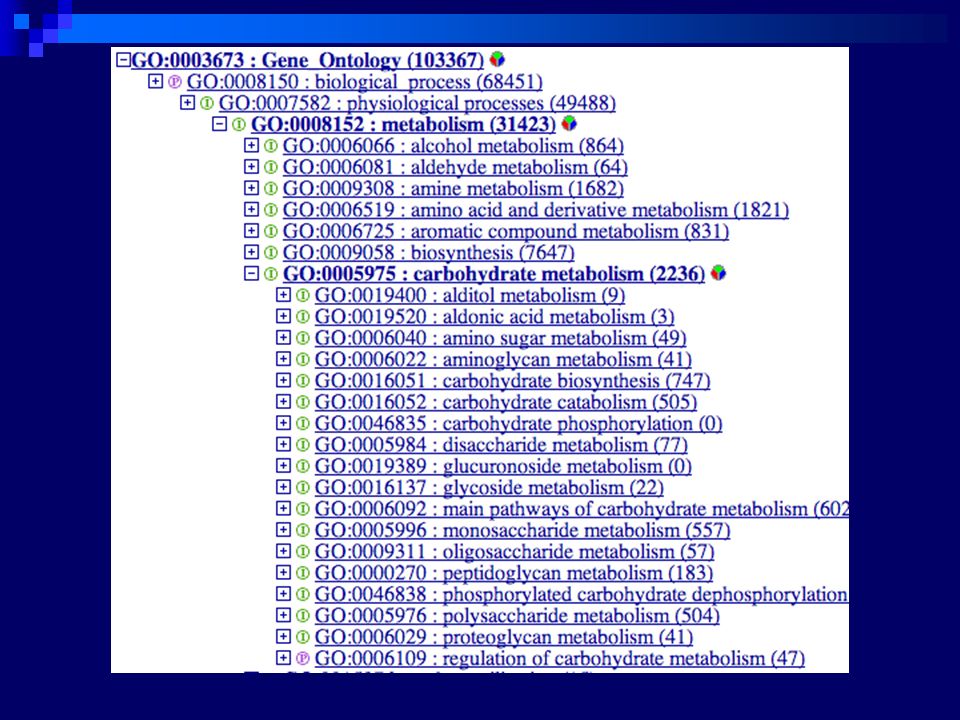
Busca com a palavra “collagenase”

135
Conteúdo do GO molecular function 7422 termos
biological process termos cellular component termos all ,866 terms

Apresentações semelhantes