Carregar apresentação
A apresentação está carregando. Por favor, espere

1
porfirias Paola Cristina Vieira R. Passos R1
Serviço de dermatologia HUEC

2
PORFIRIAS Grupo de doenças causadas por anormalidades herdadas ou adquiridas de enzimas que intervêm na biossíntese do heme levando ao acúmulo de porfirinas. Heme: composto tetrapirrólico Protoporfirina + ferro ferroso = Heme

3
Biossíntese do Heme Longa e complexa cadeia de reações
bioquímicas que tem como catalisadores diversas enzimas e resultado final o heme. 85% síntese na M.Óssea Pouco menos de 15% síntese no Fígado Inicio e final da síntese ocorrem na mitocondria e o restante no citosol

4
Biossintese do Heme

5
heme Constituinte celular essencial para vários processos metabólicos.
Habilidade única de captar e liberar oxigênio e de facilitar o transporte de elétrons. É o grupo prostético (núcleo molecular) para um grupo significativo de proteínas celulares importantes; hemoglobina, mioglobina, citocromos mitocondriais, citocromos microssomais (P450), catalase, peroxidase, prostaglandinas e outras.
 para. um grupo significativo de proteínas celulares importantes; hemoglobina, mioglobina, citocromos mitocondriais, citocromos microssomais (P450), catalase, peroxidase, prostaglandinas e outras.")
6
Porfirinas Intermediários da via do heme
Porfirinas são classificadas em 2 grupos: eritropoiética e hepática, com base no principal local do defeito enzimático específico. São diferenciadas medindo os precursores do heme na urina, nas fezes, nos eritrócitos e no plasma.

7
Tipos de Porfirias 1. Eritropoiéticas
Porfiria eritropoiética congênita Protoporfiria eritropoiética Copoporfiria eritropoiética 2. Hepato-eritrocítica

8
Tipos de Porfirias 2. Hepáticas - Porfiria aguda intermitente
Porfiria cutânea tardia Porfiria variegata ou mista Copoporfiria hereditária Porfiria por deficiência de ALA dehidratase

9
Fisiopatologia Fotossensibilidade
Porfirinas absorvem luz (bandas Soret nm) Moléculas em estado excitado Reagem com moléculas biológicas Liberação da energia sob a forma de fluorescência, fosforecência e calor Transferência de energia a moléculas de oxigênio
 Moléculas em estado excitado. Reagem com moléculas biológicas. Liberação da energia sob a forma de fluorescência, fosforecência e calor. Transferência de energia a moléculas de oxigênio.")
10
Fisiopatologia Espécies reativas de O2 Dano tecidual (peroxidação de lipídios e alterações nas membranas celulares) Liberação de mediadores e enzimas a partir de células como mastócitos e PMNL(resposta inflamatória) Síntese de colágeno (incubação de fibroblastos)
 Liberação de mediadores e enzimas a partir de células como mastócitos e PMNL(resposta inflamatória) Síntese de colágeno (incubação de fibroblastos)")
12
Porfiria eritropoiética congênita (pec)
Doença de Günther – AR Rara / Grave Inicio na infância/primeira década Forma mais frequente em crianças Mutação no gene da Uroporfinogênio III cossintetase – crom 10 Aumento da URO I e COPRO I urinárias (diagn) Maior expressão de COPRO I nas fezes (diagn) Aumento de URO I e PROTO nas hemácias e plasma
 Maior expressão de COPRO I nas fezes (diagn) Aumento de URO I e PROTO nas hemácias e plasma.")
13
PEC Manifestações Clínicas:
.Fotossensibilidade moderada-grave, bolhas,úlceras, cicatrizes em áreas expostas .Deformidades mutilantes (acral) .Infecção secundária .Cictrizes, Alopécia cicatricial .Onicólise, coiloniquia, melanoníquia .Hiper ou hipopigmentação e hipertricose facial .Esplenomegalia, anemia hemolítica, trombocitopênia .Dentes e urina de cor vermelha (fluorescência)
 .Infecção secundária. .Cictrizes, Alopécia cicatricial. .Onicólise, coiloniquia, melanoníquia. .Hiper ou hipopigmentação e hipertricose facial. .Esplenomegalia, anemia hemolítica, trombocitopênia. .Dentes e urina de cor vermelha (fluorescência)")
15
PEC AP: Clivagem subepidérmica com infiltrado inflamatório discreto
Pode haver espessamento dos feixes colágenos nas aréas cicatriciais. DD: Demais formas de porfiria, xeroderma pigmentosos, epidermólise bolhosa, hidroa vaciniforme e penfigóide bolhoso.

16
PEC Tratamento: Medidas profiláticas - cuidados com a anemia, infecção secundária, fotoproteção (física) Beta-caroteno mg dia/ PUVA Esplenectomia Transplante de MO

17
Protoporfiria Eritropoiética (ppe)
AD Rara Inicio: infância (1-4 anos) Mutação no crom 18 Deficiência da enzima ferroquetalase nos eritrocitos e fibroblastos da pele Aumento das protoporfirinas nos eritrócitos, fezes e plasma (diagn) Ausência de excreção urinária(insolúvel em água) (diagn) Fenômeno fotodinâmico levando a dano tissular
 Mutação no crom 18. Deficiência da enzima ferroquetalase nos eritrocitos e fibroblastos da pele. Aumento das protoporfirinas nos eritrócitos, fezes e plasma (diagn) Ausência de excreção urinária(insolúvel em água) (diagn) Fenômeno fotodinâmico levando a dano tissular.")
18
PPE Manifestações Clínicas
.Graus variáveis de lesões cutâneas (nariz, malares, dorso das mãos): prurido, queimação, eritema, edema, lesões urticariformes, raramente púrpuras, lesões cicatriciais, atrofias, espessamento céreo da face e dorso das mãos (metacarpofalangeana e interfalangeana), rugas periorais. Raramente bolhas. .Fotoonicólise .Anemia, calcúlo renal, colelitíase, alterações hepáticas (até cirrose e insuficiência)
: prurido, queimação, eritema, edema, lesões urticariformes, raramente púrpuras, lesões cicatriciais, atrofias, espessamento céreo da face e dorso das mãos (metacarpofalangeana e interfalangeana), rugas periorais. Raramente bolhas. .Fotoonicólise. .Anemia, calcúlo renal, colelitíase, alterações hepáticas (até cirrose e insuficiência)")
19
PpE AP: Vesícula subepidérmica e acúmulo de material homogênio eosinofilico, amorfo, PAS positivo em torno dos vasos da derme papilar. A histoquímica revela glicoproteínas, mucopolissacarídes ácidos e lípides nesse material. (≈ lipoidoproteinose) IF: depósito de IgG DD: Lipoidoproteinose, hidroa vaciniforme, erupção polimorfa à luz, urticária solar, queimadura solar e outras porfirias.
 IF: depósito de IgG. DD: Lipoidoproteinose, hidroa vaciniforme, erupção polimorfa à luz, urticária solar, queimadura solar e outras porfirias.")
22
PPE Tratamento: .Fotoproteção .Betacaroteno via oral (60-180mg/dia)
.Sugeridos sem comprovação: Drogas antimaláricas Colestiramina Vit C e E

23
Copoporfiria eritropoiética (cpe)
Muito rara (apenas 3 casos descritos) Lesões semelhantes a PPE Aumento da coproporfirina III nas hemácias Excreção urinária e fecal de porfirinas normal
 Lesões semelhantes a PPE. Aumento da coproporfirina III nas hemácias. Excreção urinária e fecal de porfirinas normal.")
24
porfiria aguda intermitente OU Porfiria hepática aguda (pai)
AD Rara (1,5: ); mais comum Ecandinávia e Lapônia Início: anos Deficiência de uroporfobilinigênio I sintetase Aumento da ALA sintetase Precursores porfirínicos ALA e PBG não fotossensibilizante Fotossensibilidade: ausente Reação cutânea: nenhuma
; mais comum Ecandinávia e Lapônia. Início: anos. Deficiência de uroporfobilinigênio I sintetase. Aumento da ALA sintetase. Precursores porfirínicos ALA e PBG não fotossensibilizante. Fotossensibilidade: ausente. Reação cutânea: nenhuma.")
25
PAI Lesão no sistema nervoso central, periférico e autonômico
Dor Abdominal, sintomas neurológicos(convulsão, dor, fraqueza, paralisia) e psiquátricos. Manifestação induzida por drogas ou álcool Aumento de ALA e PBG na urina (diagn) Ausência de tratamento específico; infusão de glicose (2l/dia a 20%), hematina ( g/dia) Retirada de drogas agravantes
 e psiquátricos. Manifestação induzida por drogas ou álcool. Aumento de ALA e PBG na urina (diagn) Ausência de tratamento específico; infusão de glicose (2l/dia a 20%), hematina ( g/dia) Retirada de drogas agravantes.")
26
Porfiria Hepática crônica ou Porfiria cutânea Tardia (PCT)
Formas: .Hereditária AD – acomete jovens .Adquirida – acomete adultos(>40 anos) Influência genética, mas desencadeada por drogas, álcool, infecções virais (Hep C e HIV) Forma mais comum de porfiria Predomina em homens Ocorrência universal
 Influência genética, mas desencadeada por drogas, álcool, infecções virais (Hep C e HIV) Forma mais comum de porfiria. Predomina em homens. Ocorrência universal.")
27
pct Forma hereditária ou tipo II
.AD - Cromossomo 1p34 (baixa penetrância) .Deficiência de uroporfobilinogênio decarboxilase (↓50%) em todos os tecidos .Excreção aumentada de uroporfirinas I e III e copoporfirinas na urina e de isocoproporfirinas na fezes .A maioria necessita de uma fator desencadeador para manifestar a doença
 .Deficiência de uroporfobilinogênio decarboxilase (↓50%) em todos os tecidos. .Excreção aumentada de uroporfirinas I e III e copoporfirinas na urina e de isocoproporfirinas na fezes. .A maioria necessita de uma fator desencadeador para manifestar a doença.")
28
PCT Forma Adquirida ou tipo I
.Deficiência de uroporfobilinogênio apenas no fígado por: defeito genético restrito ao fígado ou exposição a substâncias químicas capazes de inibir a enzima do fígado mas não da hemácia .Algumas substâncias (p.ex. álcool, estrógenos) causam porfiria apenas em predispostos e outras (p.ex hexaclorobenzeno) em todos indivíduos expostos
 causam porfiria apenas em predispostos e outras (p.ex hexaclorobenzeno) em todos indivíduos expostos.")
29
PCT .Álcool Drogas e agentes associados à expressão clínica de PCT
Inibe algumas enzimas como a uroporfibilinogênio decarboxilase, a ferroquetalase e a ALA dehidratase Induz ALA sintase hepática em pctes com PCT Supressão da eritropoiese e absorção aumentada de ferro da dieta .Estrógenos Interferem na ALA sintetase hepática

30
PCT .Hexaclorobenzeno, Bifenis clorados, Dioxina .Ferro
Inibe a uroporfobilinogênio decarboxilase após ativação metabólica dos compostos Causam uma Porfiria Química .Ferro Inibe a uroporfobilinogênio decarboxilase Sobrecarga de ferro acompanha a PCT clínica em quase todos os casos Aumento das concentrações de ferro sérico e ferritina Associação com hemocromatose

31
PCT Associação com HIV Sinais e sintomas de PCT precedentes ou subsequentes ao diagnóstico do HIV Fisiopatologia não compreendida Prejuizo da função hepática Associação com Hepatite C Mecanismo de interação desconhecido Forte ligação entre as doenças Relatos de estudos controversos

32
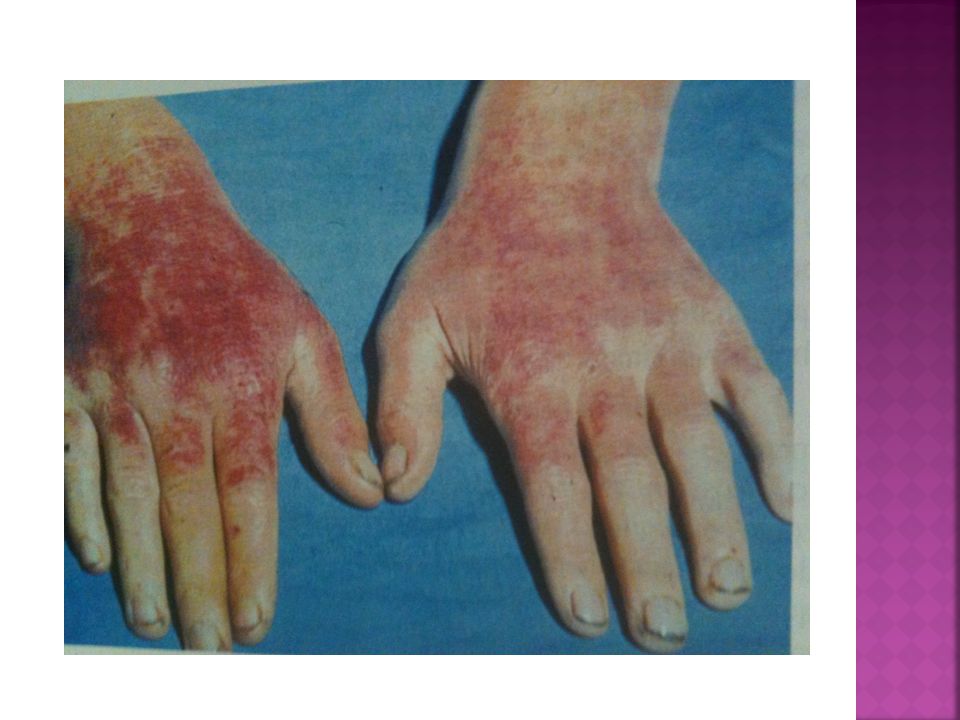
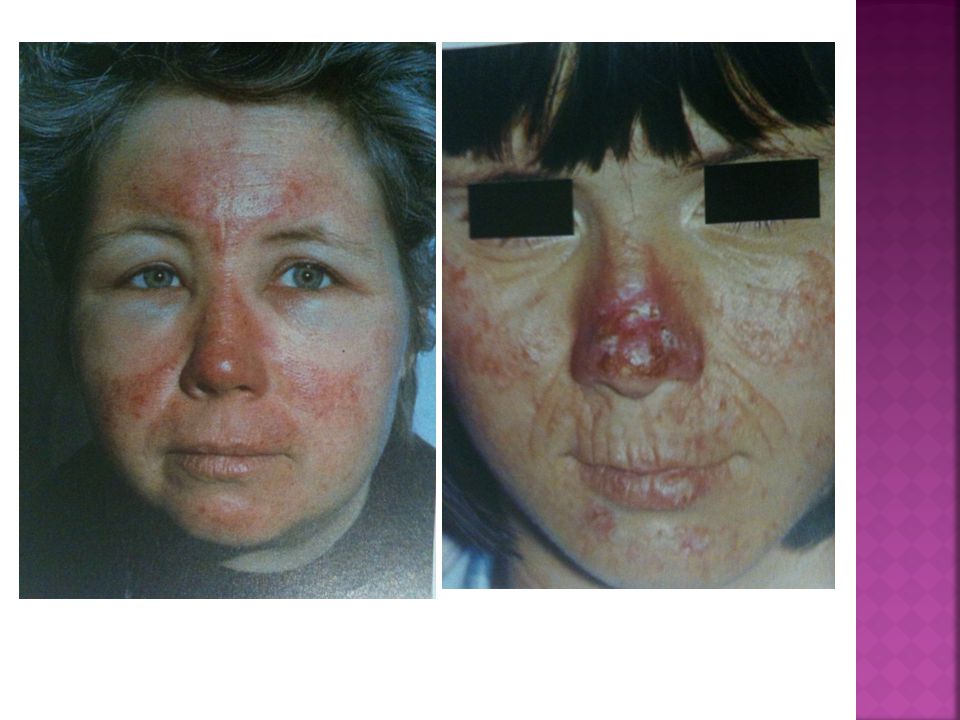
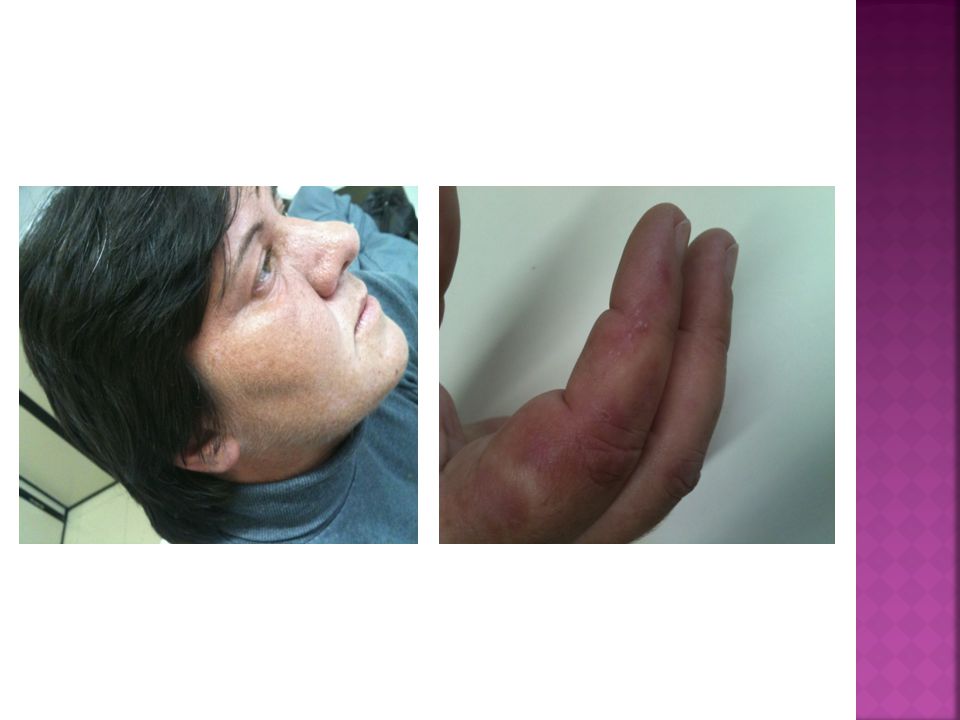
PCT Manifestações Clínicas:
.Lesões cutâneas em face, pescoço e dorso das mãos (áreas expostas); eritema, vesico-bolhas e erosões; fragilidade, cicatrizes atróficas, formação de mília, hiperpigmentação e hipopigmentação moteada, sufusão vermelho-purpúrica; hipertricose facial (região temporal e zigomática);
; eritema, vesico-bolhas e erosões; fragilidade, cicatrizes atróficas, formação de mília, hiperpigmentação e hipopigmentação moteada, sufusão vermelho-purpúrica; hipertricose facial (região temporal e zigomática);")
33
pct Porém a fotosensibilidade aguda é rara
.Urina vermelha pelo aumento da excreção de URO e COPRO porfirinas .Fluorescência vermelho-róseo à lâmpada de Wood

34
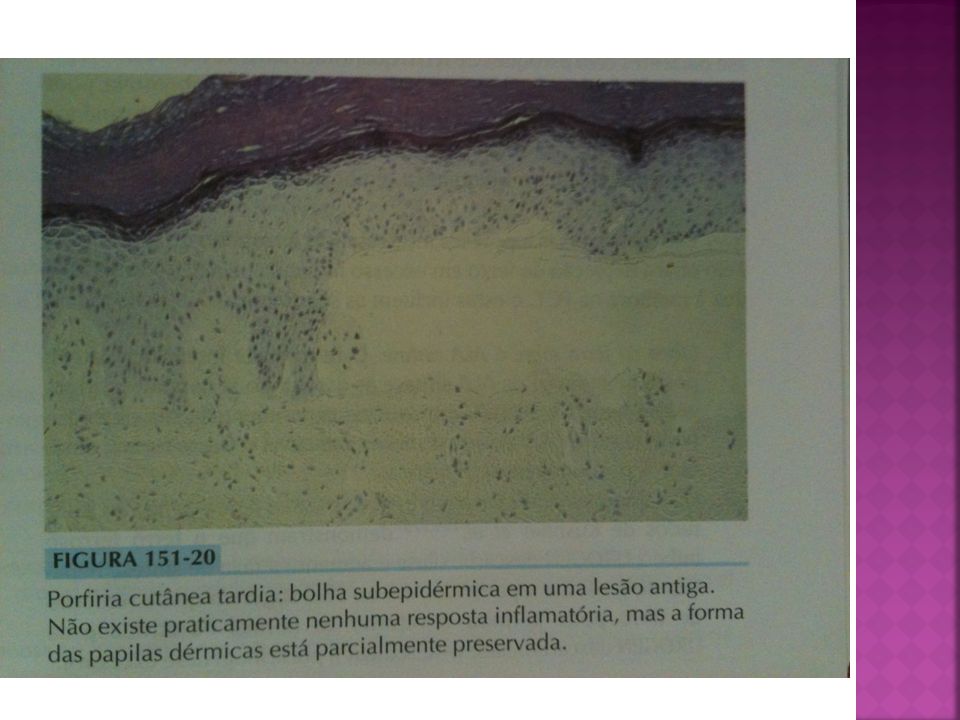
PCT AP: .Bolhas subepidérmicas caracteristicamente com base enrugada e ondulada, praticamente sem infiltrado inflamatório, e o PAS pode revelar discreto espessamento de vasos das papilas dérmicas IF: .Depósitos de IgG e C3 de padrão granular na ZMB e nas paredes vasculares

39
pct Diagnóstico: .Achados clínicos .Exame histopatológico .IFD
.Aumento das Uroporfirinas I e III e Coproporfirinas urinárias e Isocopro nas fezes .Exame da urina com a lâmpada de Wood DD Porfiria variegata, pseudoporfiria, esclerodermia e EBA

42
PCT Tratamento: .Suspensão do álcool e substâncias hepatotóxicas
.Fotoproteção .Flebotomia periódica (vol variável ~ 500ml semanais ou bisemanais) .Antimaláricos em baixas doses (Cloroquina 125mg 2x/sem ou HCQ 200mg 2x/sem)
 .Antimaláricos em baixas doses (Cloroquina 125mg 2x/sem ou HCQ 200mg 2x/sem)")
43
Porfiria variegata (PV) ou porfiria hepática mista
AD Comum na África do Sul (3/1.000) Rara em outros lugares Início: anos Deficiência de protoporfirinogênio oxidase (crom 1q22) Lesões semelhantes a PCT (+ comum homens) Manifestações de PAI (+ comum mulheres) Isoladas ou simultaneamente
 Rara em outros lugares. Início: anos. Deficiência de protoporfirinogênio oxidase (crom 1q22) Lesões semelhantes a PCT (+ comum homens) Manifestações de PAI (+ comum mulheres) Isoladas ou simultaneamente.")
44
PV Lesões cutâneas mais precoces
Acompanham-se de sintomatologia abdominal e neurológica idênticas à PAI Associação de lesões de fotossensibilidade com traumas mecânicos Fotossensibilidade mais aguda

45
pv AP: .Indistinguível da PCT DD:
.ALA e PBG urinários elevados durante os ataques agudos porém caem a níveis normais entre os ataques. Enquanto na PAI os níveis são constantemente elevados .O mesmo ocorre com os níveis fecais de Proto e Coproporfirinas

46
PV .Excreção fecal maior de Copro e menor de Uroporfirinas ao contrário da PCT . Contudo a excreção fecal total de porfirinas é muito maior na PAI em relação a PCT Tratamento: .Evitar uso de drogas desencadeantes .Proteção solar .Antimaláricos e flebotomia não efetivos

47
Coproporfiria hereditária (CPH)
AD Rara Início: anos Deficiência de coproporfirinogênio oxidase (crom 3q12) Manifestações sistêmicas semelhantes a PAI (crises abdominais, neurológicas e psiquiátricas) 20-30% apresentam fotossensibilidade semelhante a PCT e PV
 Manifestações sistêmicas semelhantes a PAI (crises abdominais, neurológicas e psiquiátricas) 20-30% apresentam fotossensibilidade semelhante a PCT e PV.")
48
Cph Níveis elevados de Coproporfirina III nas fezes e urina
Nenhum padrão histológico relatado até o momento O mesmo tratamento mencionado para PAI

49
Porfiria hepato-eritrocitária (PHE)
Profunda deficiência de Uroporfobilinogênio- descarboxilase Forma homozigótica de PCT II ou heterozigoto composto Manifestações precoces (1̊ ano de vida) Simulam PEC Urina escura é o sinal mais observado Fotossensibilidade intensa que diminui ao longo do tempo; vésico-bolhas e prurido, hipertricose, hiperpigmentação, lesões esclerodermiformes.
 Simulam PEC. Urina escura é o sinal mais observado. Fotossensibilidade intensa que diminui ao longo do tempo; vésico-bolhas e prurido, hipertricose, hiperpigmentação, lesões esclerodermiformes.")
50
phe Pode haver acometimento ocular (ectrópio), anemia e esplenomegalia
Não há evidência de ação desencadeante de droga AP mostra bolhas subepidérmicas semelhantes à PCT Fotoproteção cautelosa

51
Porfiria por deficiência de ALA sintetase
Tipo extremamente raro (<10 casos) AR Qualquer idade Atividade da ALA sintetase eritrocítica < 5% do normal Sintomas similares à PAI Não há lesões cutâneas Tratamento sintomático
 AR. Qualquer idade. Atividade da ALA sintetase eritrocítica < 5% do normal. Sintomas similares à PAI. Não há lesões cutâneas. Tratamento sintomático.")
52
Referências: Tratado de Dermatologia – Fitzpatrick Cap 151 Dermatologia – Sampaio e Rivitti Cap 60 Dermatologia Clínica – Thomas P Habif Cap 19

Apresentações semelhantes









 Defeito genético do metabolismo que causa um aumento na produção.>")










, é transmitida de pessoa a pessoa, através da água e de alimentos contaminados com matéria fecal .EM.>")




