Carregar apresentação
A apresentação está carregando. Por favor, espere

1
PORFIRIAS

2
PORFIRIAS As porfírias são um grupo de doenças em sua maioria hereditárias, ocasionadas por distúrbios enzimáticos na biossíntese do heme, no fígado ou na medula óssea, com o acúmulo de porfirinas e seus precursores

3
PORFIRIA Os porfirinogênios não são pigmentados e não fluorescem, mas são rapidamente oxidados às porfirinas correspondentes. Estes são pigmentos que exibem fluorescência vermelha, absorvendo energia principalmente na faixa de 400 a 410nm e em menor intensidade entre 500 e 600nM

4
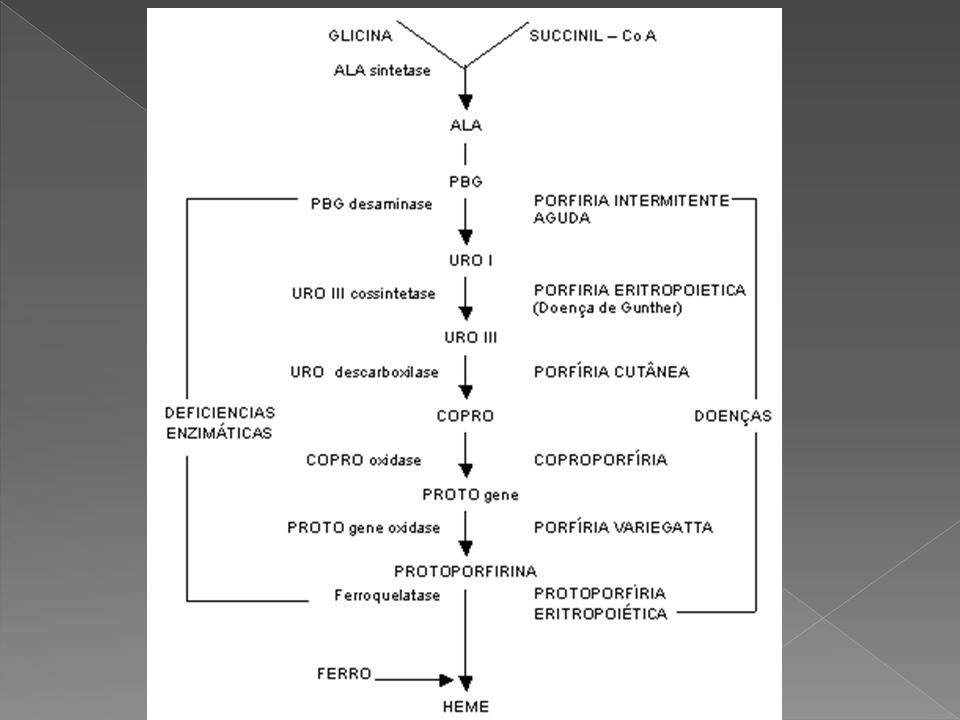
PORFIRIAS Erros Inatos do Metabolismo.
Grupo de distúrbios causados por anormalidades da síntese do heme. Podem ser genéticas ou adquiridas. São provocadas por deficiências das enzimas implicadas na síntese do heme e que levam à superprodução e acumulo de porfirinas. A deficiência de cada uma das oito enzimas necessárias à produção do Heme produz um tipo particular de Porfiria

5
PORFIRIA O reconhecimento do acúmulo e da excreção aumentada destes compostos, em doenças metabólicas, data de 1874 quando do relato de um caso com sinais e sintomas sugestivos de porfíria eritropoiética congênita. Os legendários lobisomens e os vampiros

6
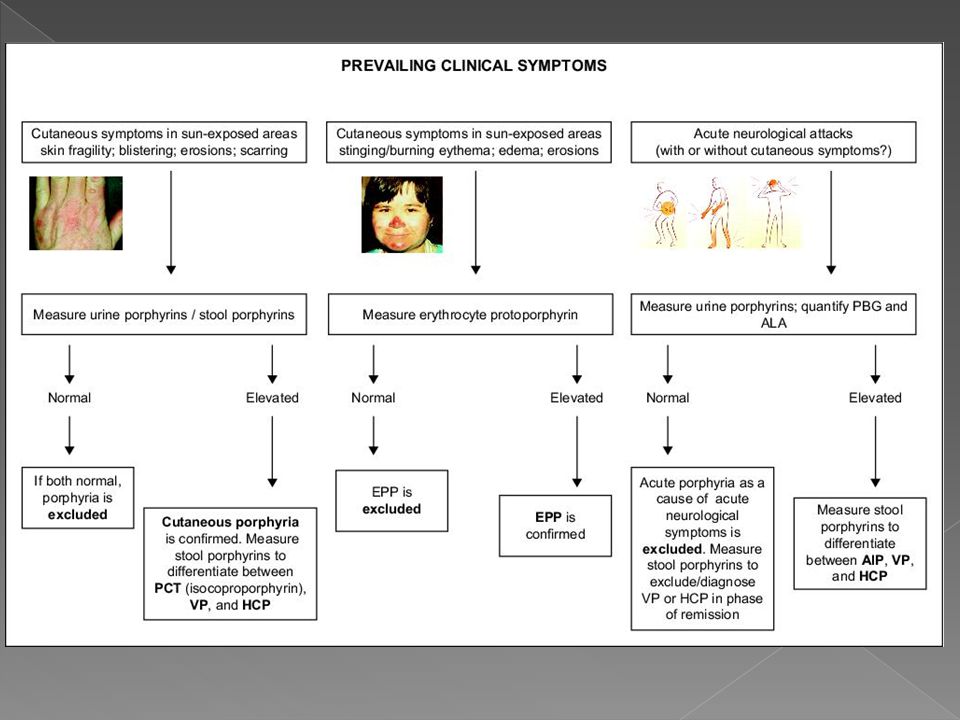
PORFIRIA Cada forma clínica de porfíria encontra-se associada a um determinado defeito enzimático na biossíntese do heme

9
PORFIRIA A Porfíria Aguda Intermitente (P.A.I.) tem sido a mais estudada a nível molecular. Geneticamente heterogênea, possui pelo menos quatro genes mutantes distintos para a Porfobilinogênio deaminase, sendo que dois caracterizam-se pela diminuição da atividade enzimática e os outros dois pela produção de proteína enzimática alterada.

10
PORFIRIA FOTODINÂMICA E MANIFESTAÇÕES CLÍNICAS
As porfirinas depositadas na pele ou circulando nos vasos sangüíneos da derme são ativadas pela luz ultravioleta de ondas longas principalmente na faixa de Soret ( nm), capazes de penetrar através de vidros comuns. As moléculas das porfirinas excitadas reagem desta forma diretamente com oxigênio molecular formando peróxidos que liberam O++ e lesam tecidos oxidando membranas lipídicas, reagindo com proteínas, oxidando ácidos nucléicos, interferindo nas funções das organelas celulares e ativando complemento no soro.
, capazes de penetrar através de vidros comuns. As moléculas das porfirinas excitadas reagem desta forma diretamente com oxigênio molecular formando peróxidos que liberam O++ e lesam tecidos oxidando membranas lipídicas, reagindo com proteínas, oxidando ácidos nucléicos, interferindo nas funções das organelas celulares e ativando complemento no soro.")
12
PORFIRIA FOTODINÂMICA E MANIFESTAÇÕES CLÍNICAS
Os diferentes sintomas das Porfirias, entretanto, podem estar relacionados às propriedades físico-químicas de cada porfirina. Em modelo experimental, por exemplo, as Protoporfirinas acumulam-se predominantemente nas mitocôndrias e em membranas biológicas, enquanto que as uroporfirinas acumulam-se predominantemente em lisossomos. Isto explicaria em parte os sintomas de fotossensibilidade aguda com dor e eritema nas Protoporfírias e a evolução crônica, marcada pela fragilidade cutânea, na Porfíria Cutânea Tarda.

14
PORFIRIA Fotodinâmica e manifestações clínicas
A dor abdominal é considerada um dos sintomas neurológicos mais comuns nas porfírias, por comprometimento do sistema nervoso autônomo. Entretanto, a patogenia destas manifestações não está esclarecida. Sabe-se que o ALA age como uma neurotoxina e que a deficiência do heme poderia alterar nas funções neurológicas de forma direta e indireta (provocando no fígado aumento de triptofano e 5-hidroxitriptofano).
.")
15
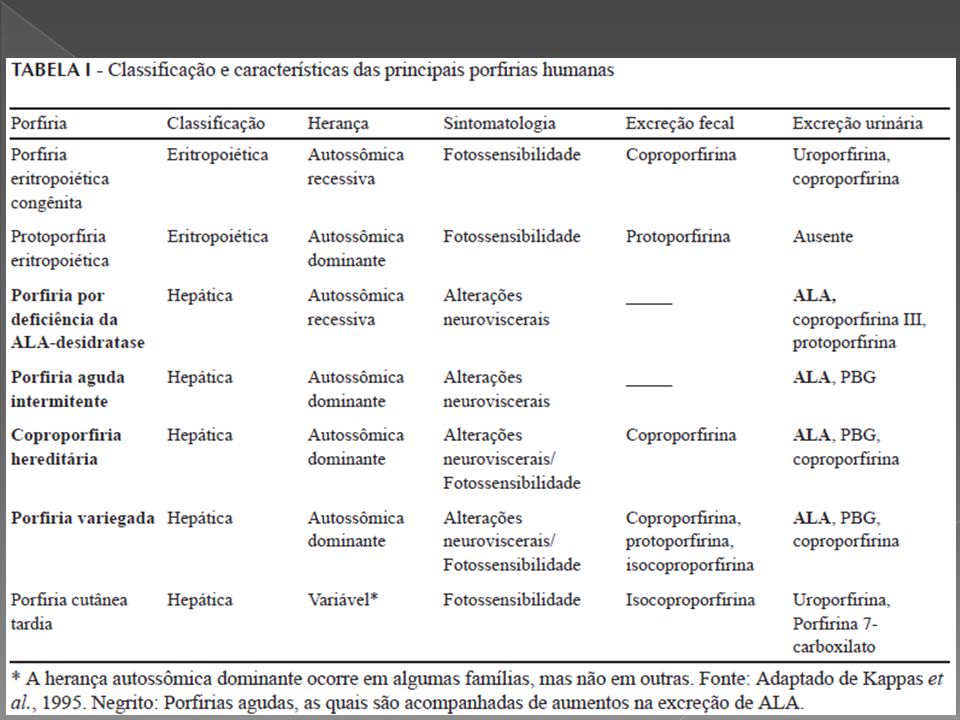
CLASSIFICAÇÃO DAS PORFIRIAS
PORFIRIAS ERITROPOIÉTICAS – Não aguda 1. Porfiria eritropoiética congênita 2. Protoporfiria eritropoiética 3. Coproporfiria eritropoiética PORFIRIAS HEPÁTICAS - AGUDAS 1. Porfiria aguda intermitente (PAI) 2. Porfiria variegata ou mista 3. Coproporfiria hereditária 4. Porfiria por deficiência de ALA deidratase PORFIRIA HEPÁTICA – NÃO AGUDA 1. Porfiria cutânea tardia (PCT) PORFIRIA HEPATOERITROPOIÉTICA
 2. Porfiria variegata ou mista. 3. Coproporfiria hereditária. 4. Porfiria por deficiência de ALA deidratase. PORFIRIA HEPÁTICA – NÃO AGUDA. 1. Porfiria cutânea tardia (PCT) PORFIRIA HEPATOERITROPOIÉTICA.")
16
PORFIRIAS Classificação
Tradicionalmente as Porfírias têm sido classificadas em dois grandes grupos, tendo por base o local de maior produção de porfirinas- Porfirias Eritropoiéticas e Porfírias Hepáticas. No entanto, evidências de superprodução de porfirinas em ambos os tecidos tornam esta sistemática insatisfatória, mantendo-se sua utilização por motivos didáticos

17
PORFIRIA

19
PORFIRIAS Classificação
Recentemente, Cripp sugere que as Porfírias sejam convenientemente agrupadas de acordo com os sintomas apresentados

20
PORFIRIA

21
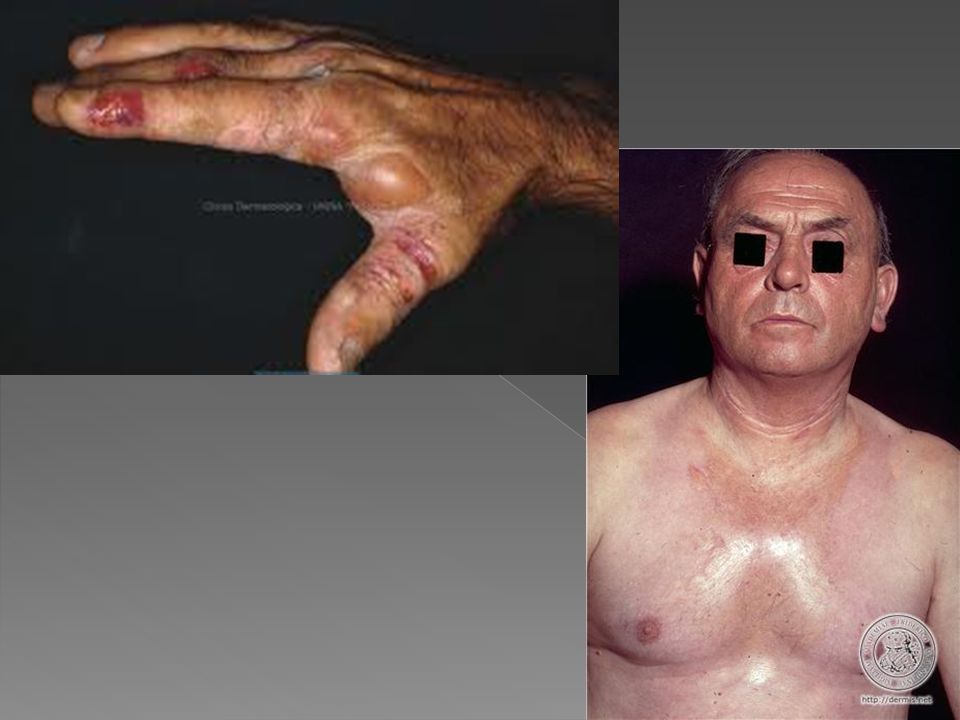
PORFÍRIA ERITROPOIÉTICA CONGÊNITA (DOENÇA DE GUNTHER)
Doença extremamente rara, Autossômica recessiva, Inicia-se entre dois a três anos de idade, com fotossensibilidade acentuada, urina de coloração avermelhada, esplenomegalia e anemia hemolítica.

22
PORFÍRIA ERITROPOIÉTICA CONGÊNITA (DOENÇA DE GUNTHER)
Deficiência da uroporfirinogênio III sintetase, Consequente excesso de uro e coproporfirina I na urina, plasma, fezes, eritrócitos e medula óssea- Urina vermelha

23
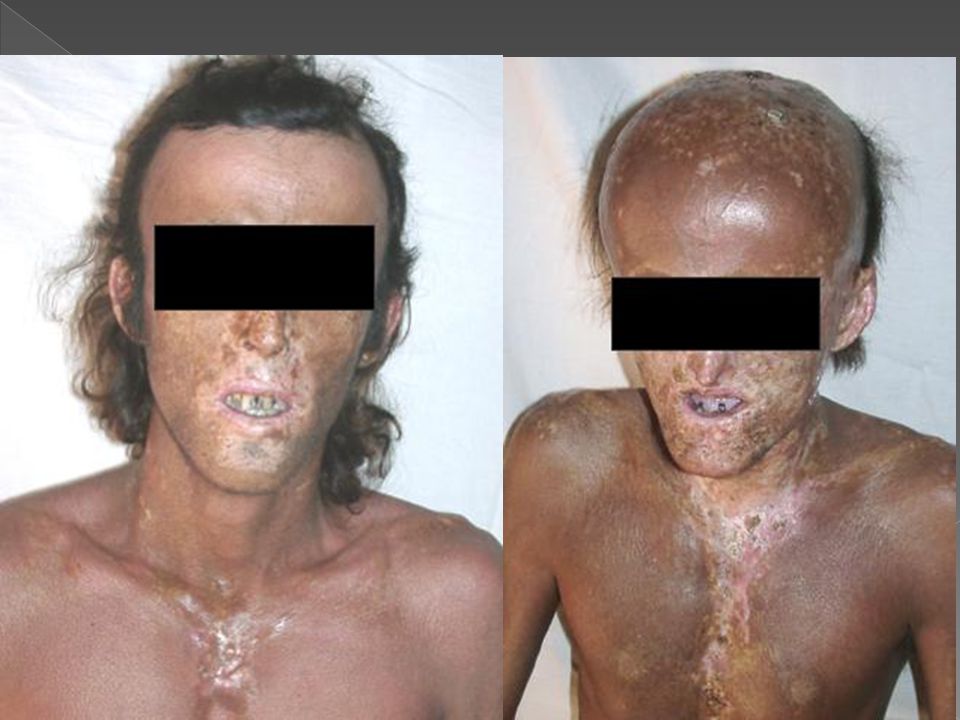
PORFÍRIA ERITROPOIÉTICA CONGÊNITA (DOENÇA DE GUNTHER)
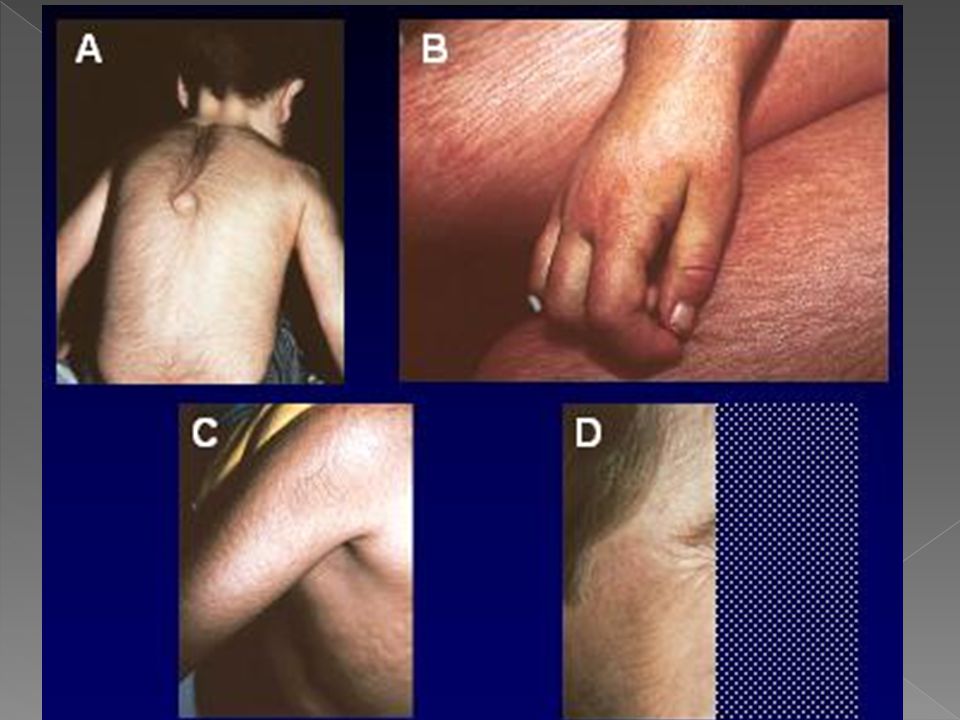
Com a lâmpada de Wood pode-se obter fluorescência dos dentes, unhas, urina, da suspensão aquosa de fezes; e com o microscópio de fluorescência pode-se observar a fluorescência vermelha dos eritrócitos em esfregação de sangue periférico. As lesões vésico-bolhosas recorrentes levam a cicatrizes retráteis, ulcerações, mutilações e deformidades dos dedos, unhas, nariz e orelhas.

25
PORFÍRIA ERITROPOIÉTICA CONGÊNITA (DOENÇA DE GUNTHER)
Conjuntivite Hipertricose na face e extremidades Ceratite Hiperpigmentação Retardo de crescimento Fragilidade óssea devido a hiperplasia da medula óssea que se insinua na cortical Aumento de reticulócitos.

27
PORFÍRIA ERITROPOIÉTICA CONGÊNITA (DOENÇA DE GUNTHER)
Esplenomegalia Anemia hemolítica Trombocitopenia Fotossensibilidade Alopecia cicatricial Onicólise Coiloníquia Melanoníquia

28
PORFÍRIA ERITROPOIÉTICA CONGÊNITA (DOENÇA DE GUNTHER)
TRATAMENTO Fotoproteção Betacaroteno VO. Nas formas graves há necessidade de hemodiálise, esplenectomia e transplante de medula óssea.

30
PORFÍRIA ERITROPOIÉTICA CONGÊNITA (DOENÇA DE GUNTHER)
Prognóstico: Muito reservado, poucos pacientes ultrapassam a quarta ou quinta décadas da vida Morte por anemia hemolítica, hemossiderose, cirrose hepática e falência hepatorrenal.

31
PORFÍRIA ERITROPOIÉTICA CONGÊNITA (DOENÇA DE GUNTHER)
")
32
PORFÍRIA ERITROPOIÉTICA CONGÊNITA (DOENÇA DE GUNTHER)
")
33
PROTOPORFIRIA ERITROPOIÉTICA
Autossômica dominante, Uma das mais freqüentes, Determinada pela deficiência da ferroquelase, enzima que acelera a incorporação do ferro na protoporfirina IX

34
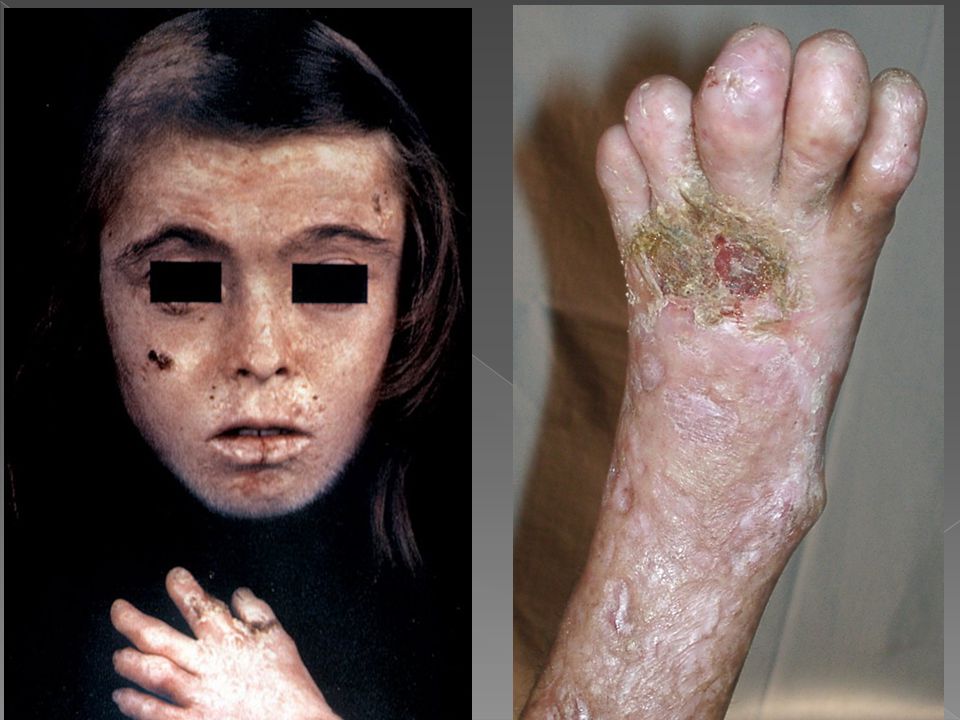
PROTOPORFIRIA ERITROPOIÉTICA
Manifesta-se sintomaticamente entre dois e cinco anos de idade Fotossensibilidade acentuada e dolorosa, caracterizada por eritema, ardência e prurido após curtos períodos de exposição, que podem regredir em 24 horas ou persistir com lesões papulovesiculosas por períodos mais longos Placas edematosas, eritema, urticária, púrpura, raras bolhas. Posteriormente surgem cicatrizes, alterações articulares, infiltração difusa, prurido e queimação durante a exposição solar. Evoluindo com eczematização, hipo e hiperpigmentação e cutis romboidal

36
PROTOPORFÍRIA ERITROPOIÉTICA

37
PROTOPORFÍRIA ERITROPOIÉTICA
Em alguns pacientes há hipersensibilidade que responde favoravelmente à esplenectomia. Pode ocorrer Fibrose portal e periportal com deposição de pigmento nos canalículos biliares, levando a Insuficiência hepática. Colecistite e colelitíase tem sido descritas devido a cálculos de protoporfirina IX.

39
PROTOPORFÍRIA ERITROPOIÉTICA
Há acúmulos de protoporfirinas nos eritrócitos, plasma, medula óssea, bile e fezes. Pode ser demonstrada a fluorescência dos eritrócitos por microscópio de fluorescência. Não há protoporfirina na urina devido a insolubilidade da mesma na água. Fotoproteção é mais importante. Nas crises graves: hospitalização, proteção solar total e ventilação. Pode ser utilizado betacaroteno VO, PUVA ou UVB de banda estreita de curta duração

41
COPROPORFIRIA ERITROPOIÉTICA
Muito rara, por isso necessita de maiores conhecimentos a respeito de suas alterações metabólicas. Há elevados níveis de coproporfirina nos eritrócitos e níveis mais baixos de uro e protoporfirina nos eritrócitos e nas fezes. Lesões semelhantes às da PPE Não há alterações na urina. Nas áreas expostas ha intenso edema de fotossensibilização.

42
PORFIRIA HEPATOERITROPOIÉTICA
Alterações bioquímicas de porfíria hepática e protoporfíria eritropoiética, Severa deficiência de uroporfirinogênio decarboxilase Autossômica recessiva

43
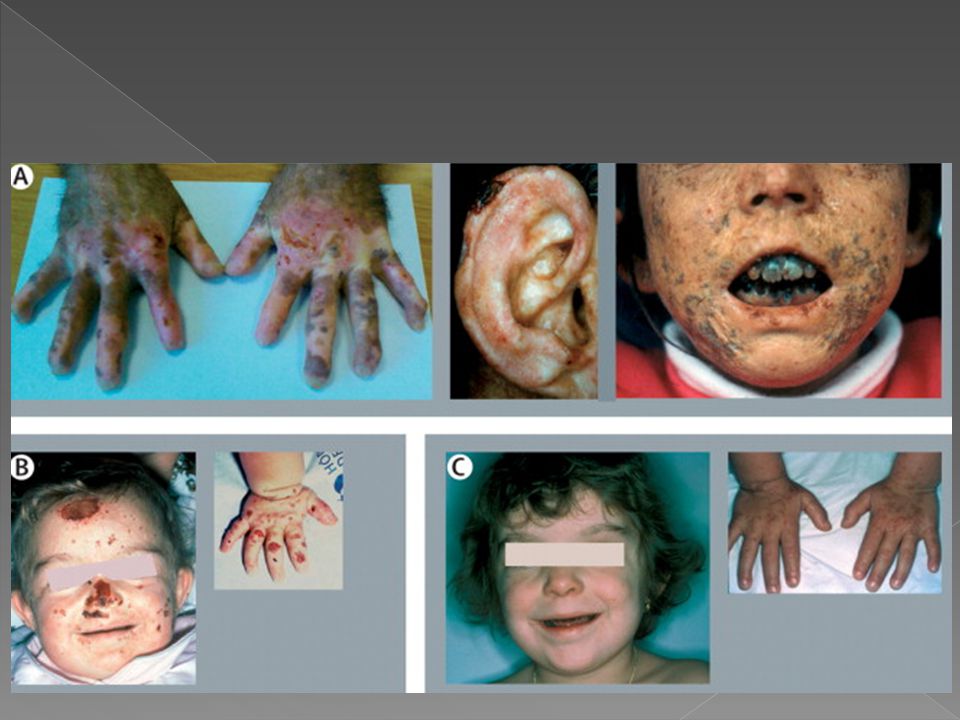
PORFIRIA HEPATOERITROPOIÉTICA
Os sintomas iniciam-se na infância, em torno de cinco anos, Intensa fotossensibilidade, com vesículas em áreas expostas e fragilidade cutânea que se sobrepõem aos da doença de Gunther devido a mutilações e cicatrizes Bolhas, erosões, cicatrizes, mutilações, hipertricose. Em fases mais tardias, há cicatrizes esclerodermiformes, hiperpigmentação e deformidades mutilantes acrais

44
PORFIRIA HEPATOERITROPOIÉTICA
Eritrodontia Anemia hemolítica severa Hepatoesplenomegalia Caracteriza-se ainda pela presença de zinco-protoporfirina nos eritrócitos e isocoproporfirinas na urina e fezes. A atividade de Uroporfirinogênio decarboxilase encontra-se reduzida a cerca de 5 % a 10% do normal.

45
PORFIRIA AGUDA INTERMITENTE PORFIRIA SUECA PIRROLOPORFIRIA
É uma doença rara, Autossômica dominante, Devido a deficiência da porfobilinogênio deaminase, Caracterizando-se pelo acúmulo de ALA e porfobilinogêrnio na urina, que pode ser demonstrado pelo teste de Watson-Schwartz.

46
PORFIRIA AGUDA INTERMITENTE PORFIRIA SUECA PIRROLOPORFIRIA
Não ocorre fotossensibilização. A doença manifesta-se sempre após a puberdade, principalmente em mulheres. Ocorrem episódios de dor abdominal associada a vômito e constipação, além de paresia e paralisias periféricas e quadros de psiconeurose Não apresenta lesões cutâneas. Não há tratamento específico: hospitalização, glicose, hematina e retirada das drogas agravantes.

48
PORFIRIA AGUDA INTERMITENTE PORFIRIA SUECA PIRROLOPORFIRIA
Estas crises podem ser desencadeadas por barbitúricos, sulfonamidas, dapsona, griseofulvina, anticonvulsivantes, sulfo-niluréia e estrógenos, além do jejum e do stress. É comum a laparotomia branca nestes pacientes, bem como a confusão diagnóstica com doenças neurológicas e psiquiátricas. A urina fresca costuma ser clara, escurecendo quando deixada em repouso. Os episódios agudos podem ainda seguir qualquer doença febril, ciclos menstruais ou ocorrer durante a gravidez.

49
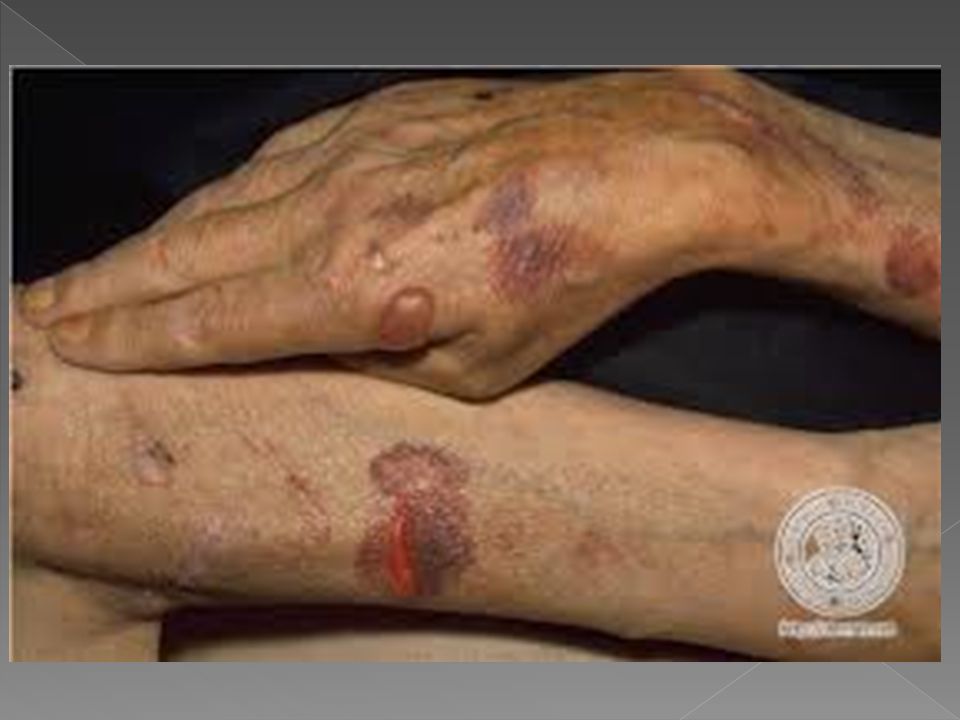
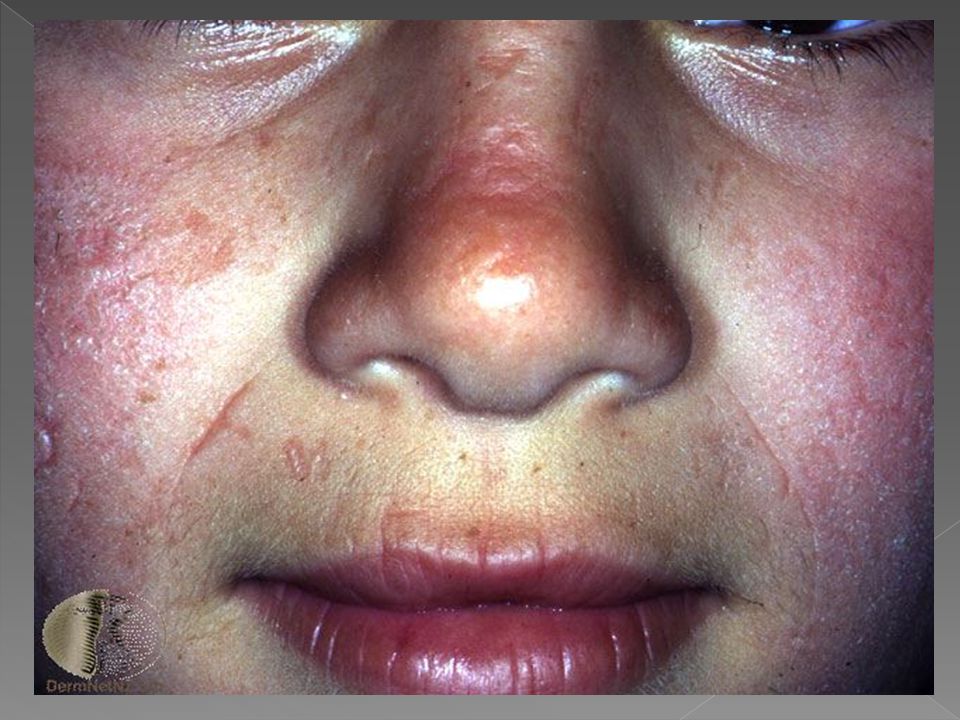
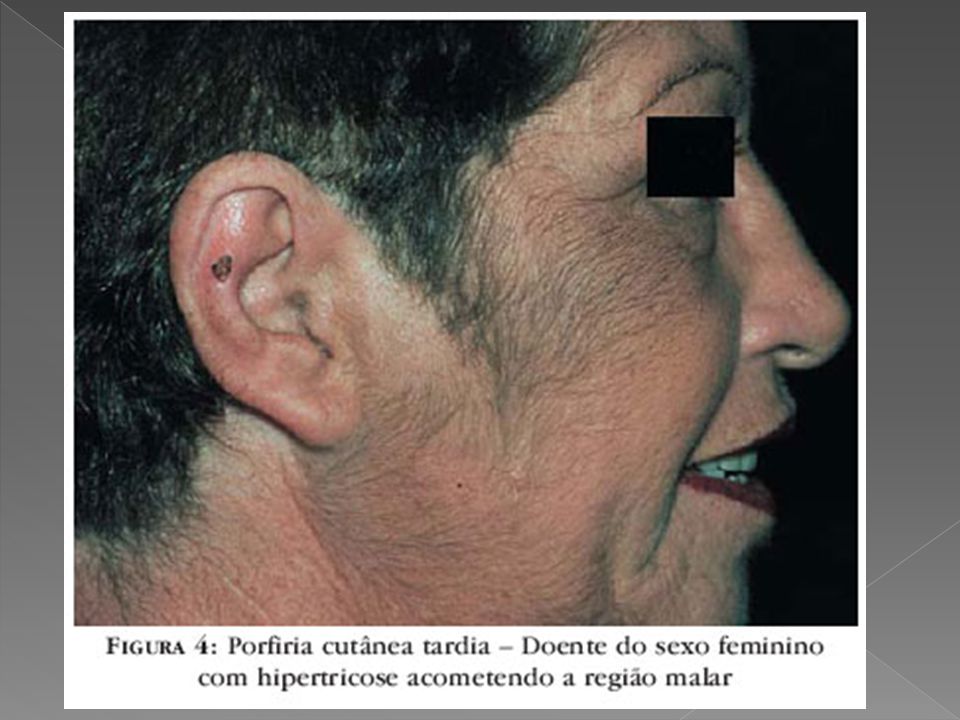
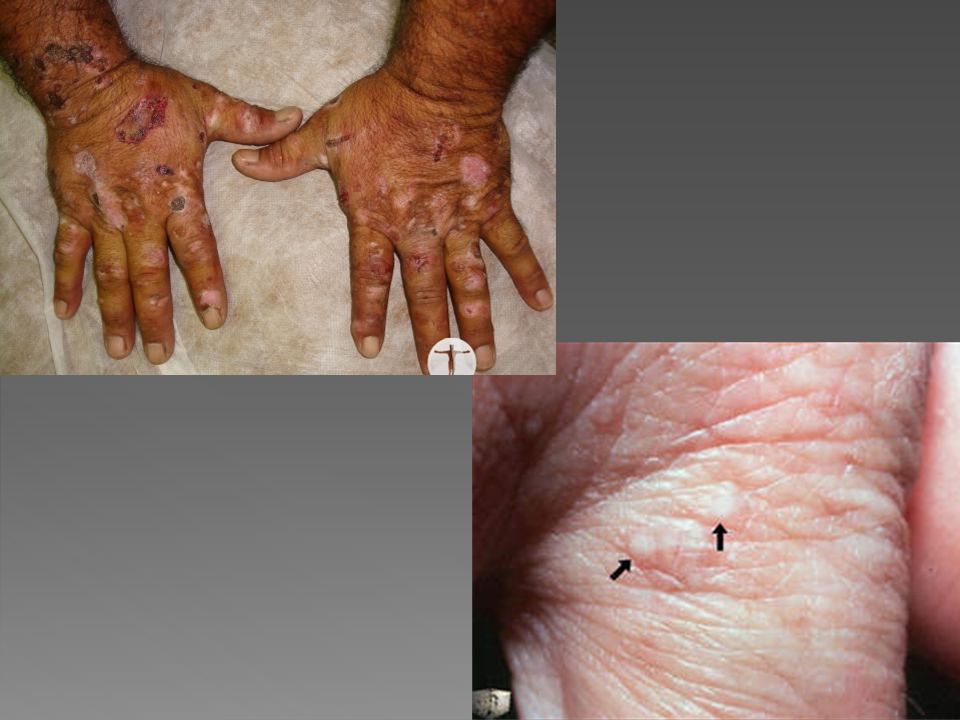
PORFIRIA CUTÂNEA TARDA
Geralmente manifesta-se em adultos entre a terceira e a quarta décadas, porém raramente ocorre em crianças, abaixo aos 15 anos. Afeta mais homens do que mulheres, É a forma mais comum de porfiria, considerada dominante, devido a deficiência da Uroporfirinogênio decarboxilase, ocorrendo eliminação de uro e coproporfirina na urina e fezes e deposição de porfirinas também na pele, com fotossensibilização e lesões cutâneas.

51
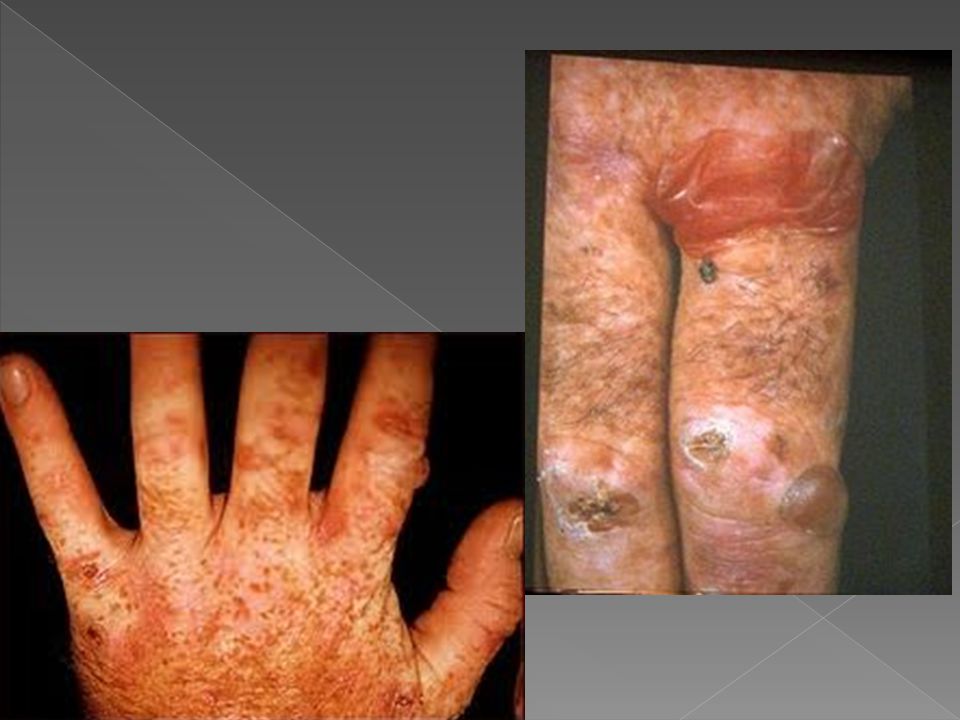
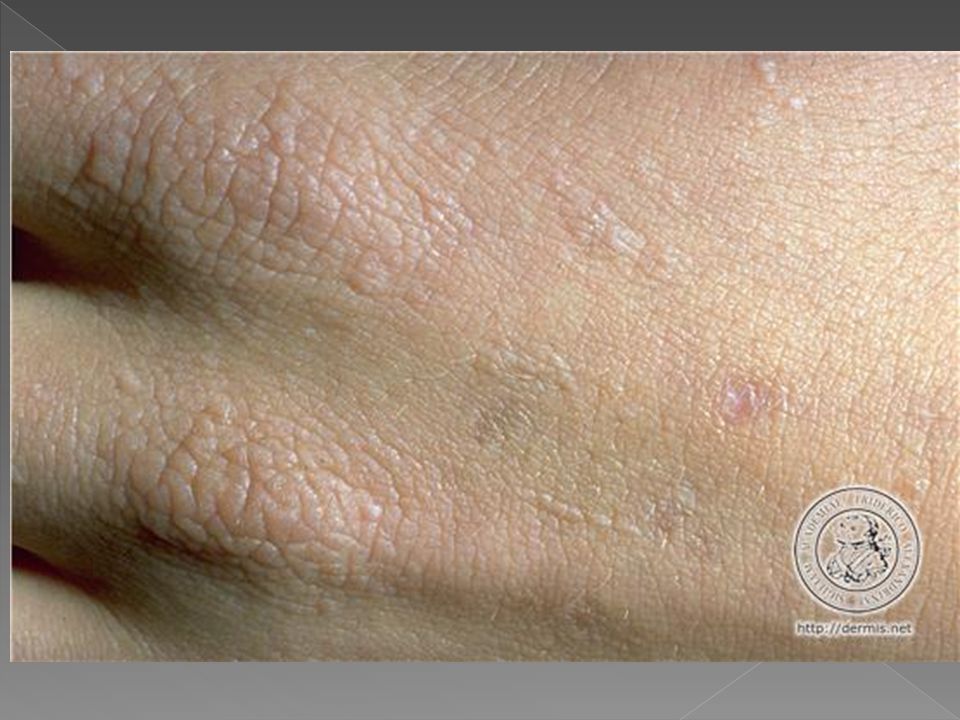
PORFIRIA CUTÂNEA TARDA
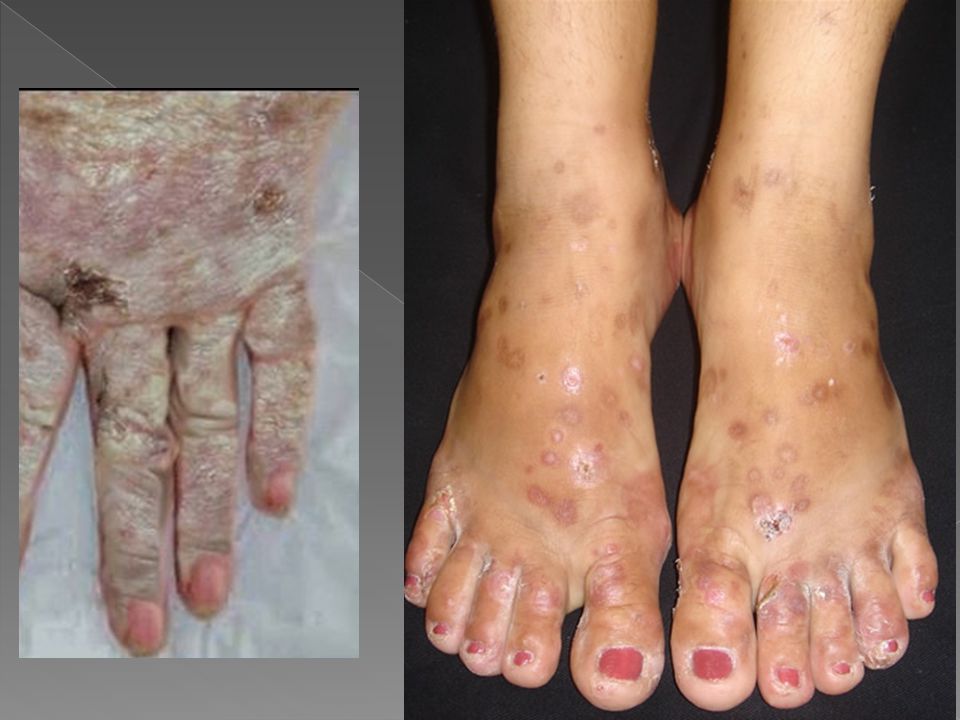
Vesículas e bolhas principalmente no dorso das mãos e demais áreas expostas Erosões e ulcerações Fragilidade cutânea Hipertricose Melanose. Cronicamente ocorrem ainda mília, alterações esclerodermiformes, áreas de atrofia e alterações ungueais como coiloníquia e estrias longitudinais pigmentadas

53
PORFIRIA CUTÂNEA TARDA-TIPOS
Forma adquirida ou tipo I Mais comum (75% dos casos). Acomete adultos maiores de 40 anos. Decorre da deficiência de uroporfobilinogênio decarboxilase no fígado. Principais fatores de risco para o desenvolvimento de PCT: hemocromatose genética subclínica, hepatite C, álcool e estrógeno (anticoncepcional oral, terapia de reposição hormonal oral).
. Acomete adultos maiores de 40 anos. Decorre da deficiência de uroporfobilinogênio decarboxilase no fígado. Principais fatores de risco para o desenvolvimento de PCT: hemocromatose genética subclínica, hepatite C, álcool e estrógeno (anticoncepcional oral, terapia de reposição hormonal oral).")
54
PORFIRIA CUTÂNEA TARDA-TIPOS
Tipo hereditários ou tipo II Autossômica dominante com baixa penetração é mais comum em jovens. História familiar em menos de 7%. Lesões cutâneas: eritema, vesico-bolhas e erosões após exposição solar. Aumento da fragilidade cutânea na pele fotoexposta, principalmente no dorso das mãos e antebraços. Traumas mínimos laceram a pele, deixando erosões bem delimitadas. Ocorre bolhas de até 1cm de diâmetro e dolorosas que evoluem com crostas e resolvem-se em algumas semanas, deixando cicatrizes atróficas, milio e hiper ou hipocromia reticulada. O acúmulo de porfirina é carcinogênico para o fígado, havendo risco adicional de carcinoma hepatocelular. A urina é vermelha.

56
PORFIRIA CUTÂNEA TARDA
É basicamente uma doença hepática, podendo ser também precipitada por cirrose alcoólica, por estrógenos, pela ingestão de hexaclorobenzeno, pelo álcool e por cirrose devido a arsenicalismo crônico. Associação com diabetes em cerca de 25 a 50% aos casos. Doses mais elevadas de cloroquina podem provocar exacerbações transitórias com febre, vômitos e piora da função hepática, seguidas de remissão.

58
PORFIRIA VARIEGATA Autossômica dominante, por deficiência da Protoporfirinogênio oxidase, representa uma combinação entre a Aguda Intermitente e a Cutânea Tarda. Inicia-se sempre após a puberdade, geralmente entre a quarta e quinta décadas. Ocorrem manifestações sistêmicas como dores abdominais, sintomas neurológicos centrais ou periféricos e distúrbios psiquiátricos, além de sintomas cutâneos por fotossensibilização (vesículas, hipertricose, mília, cicatrizes e cutis romboidalis).
.")
59
PORFIRIA VARIEGATA Os pacientes podem apresentar apenas sintomas cutâneos ou apenas manifestações sistêmicas ou ambos. Ocorrem surtos agudos como na Porfiria Aguda Intermitente e a exposição a drogas como sulfa, griseofulvina e barbitúricos, bem como situações de stress, podem desencadear crises.

61
PORFIRIA VARIEGATA Há níveis elevados de ALA, PBG e coproporfirina na urina, PBG na bile e protoporfirina nas fezes. A urina apresenta fluorescência com a lâmpada de Wood e o teste para PBG com o aldeído de Ehrlich (Watson-Schwartz) é positivo.
 é positivo.")
62
COPROPORFIRIA HEREDITÁRIA
Trata-se de uma doença rara, autossômica dominante, que ocorre por deficiência da Coproporfirinogênio, com excesso de ALA, PBG e coproporfirina na urina e de coproporfirina nas fezes. Evolui com surtos agudos, fotossensibilização, lesões cutâneas semelhantes as da Porfiria Cutânea Tarda e sintomas de disfunção neurológica.

63
PORFIRIA POR DEFICIÊNCIA DE ALA SINTETASE
Muito rara. É autossômica dominante e acomete qualquer idade. Apresenta sintomas semelhantes à PAI.

64
PORFIRIA Histopatologia
O quadro histopatológico isoladamente não é suficiente para se firmar o diagnóstico de Porfiria.

65
PORFIRIA Histopatologia
Na derme, depósitos de substância hialina PAS-positiva-diastase-resistente, que se situam principalmente em torno de vasos capilares. Presença excessiva de colágeno tipo IV em torno de vasos. Nas lesões bolhosas a membrana PAS-positiva-diastase-resistente fica no assoalho da bolha, e a reação inflamatória e geralmente escassa na ausência de infecção secundária.

66
PORFIRIA Histopatologia
A microscopia eletrônica há reduplicação da lâmina basal aos vasos capilares na derme superior e na junção dermoepidérmica. À imunofluorescência encontra-se depósitos de IgG e em menor escala de IgM ou complemento em torno de vasos capilares na derme superior e na junção dermoepidérmica. Estas alterações são comumente encontradas na P.C.T., porém não são patognomônicas, sendo também encontradas em outras formas de Porfirias.

67
PORFIRIA Histopatologia
Na P.A.I. pode-se encontrar, no fígado, necrose centrolobular ou cirrose, além de focos de desmielinização em diversos níveis do sistema nervoso.

69
PORFIRIA Tratamento Nas formas eritropoiéticas, com severa fotossensibilização, há indicação do uso de fotoprotetores opacos contendo óxido de zinco ou dióxido de titânio. O beta-caroteno pode ser utilizado em doses que variam de 30 a 150mg/dia e seus efeitos se fazem notar em torno de um a três meses de uso. Em pacientes com insuficiência hepática pode ser utilizada a associação de colestiramina (cerca de 12g/dia) e vitamina E (100 unid./dia), com melhora clínica e bioquímica.
 e vitamina E (100 unid./dia), com melhora clínica e bioquímica.")
70
PORFIRIA Tratamento Na Porfiria Cutânea Tarda a FLEBOTOMIA tem sido utilizada a intervalos de oito a 20 dias, retirando-se entre 300 a 500ml de sangue para manter o nível de ferro sérico entre 50 a 60 microgramas/ 100ml. Os sintomas cutâneos de fotossensibilização melhoram em semanas, mas os níveis de porfirinas na urina demoram de seis a oito meses para voltar ao normal. m São necessários em geral de três a 12 sessões de flebotomia, após as quais os sintomas continuam a melhorar. O controle pode ser feito com dosagens de ferro sérico, que deve ser mantido abaixo de 60 µg/ 100ml e pela dosagem de porfirinas na urina, que devem ser mantidas abaixo de 500 µg por 24 horas

71
PORFIRIA Tratamento Constituem contra-indicação para a flebotomia a angina ou doença coronariana, miocardiopatia, insuficiência renal e lúpus eritematoso sistêmico. Nestes pacientes a cloroquina pode ser utilizada em geral na dose de 500mg/dia e depois em dias alternados ou 100 a 125 mg duas a três vezes por semana, preferencialmente a hidroxicloroquina, por estar aparentemente menos relacionada a efeitos colaterais como a retinopatia. Estes antimaláricos formam complexos hidrossolúveis com as porfirinas no fígado, que são prontamente removidos e excretados pela urina. Se a dosagem for aquela usual para o tratamento de lúpus eritematoso ocorre uma maciça mobilização de porfirinas hepáticas, que solubilizadas e excretadas pela urina provocam crises de dor abdominal, febre e elevação de transaminases.

72
PORFIRIA Tratamento Nas crises agudas das porfirias hepáticas a utilização de infusão de grande quantidade de glicose (300 a 500g/dia) pode dar excelente resultado (a glicose inibiria a ALA- sintetase). Em casos de dor abdominal discreta a administração oral pode ser suficiente. Em pacientes que não toleram a glicose pode-se usar a hematina ou o heme-arginato.
 pode dar excelente resultado (a glicose inibiria a ALA- sintetase). Em casos de dor abdominal discreta a administração oral pode ser suficiente. Em pacientes que não toleram a glicose pode-se usar a hematina ou o heme-arginato.")
73
PORFIRIA Tratamento A clorpromazina e a meperidina podem ainda ser necessárias para o alívio da dor. É de grande importância o afastamento das substâncias desencadeantes, devendo ser evitados ainda o stress e o jejum. o aconselhamento genético deve ser considerado em cada caso particularmente.

74
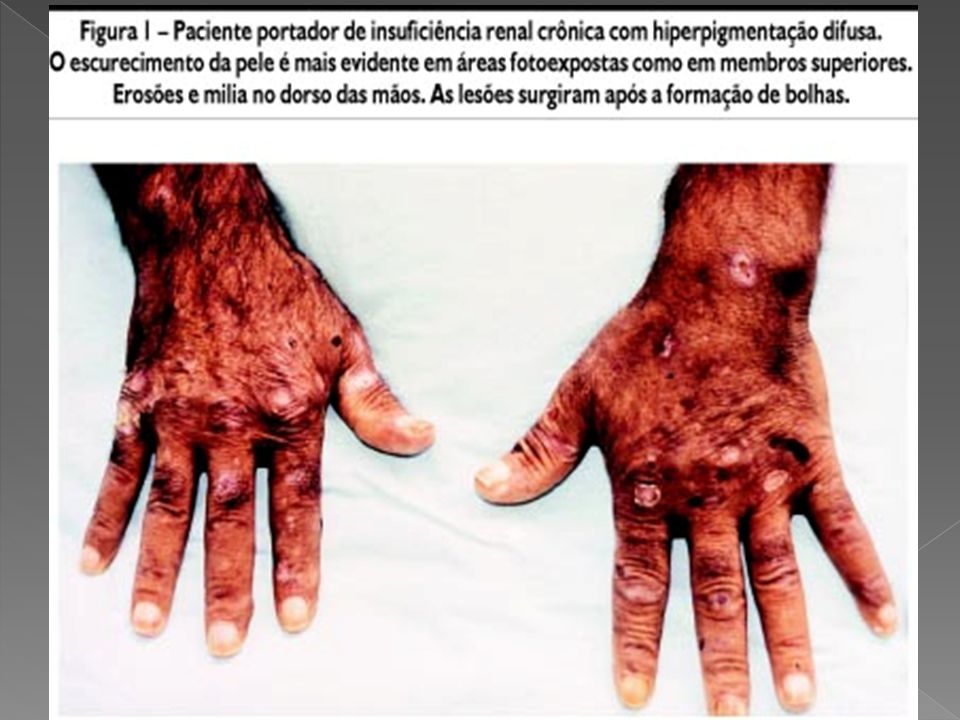
PSEUDOPORFIRIA Dermatose vesicobolhosa, causada por fototoxicidade, que acomete pacientes renais crônicos em terapia de substituição renal (diálise peritoneal ou hemodiálise) cuja clínica e alterações histopatológicas são semelhantes à porfiria cutânea tarda (hepática crônica).
 cuja clínica e alterações histopatológicas são semelhantes à porfiria cutânea tarda (hepática crônica).")
75
PSEUDOPORFIRIA Outras condições como: drogas, câmaras de bronzeamento e PUVA também foram implicadas na etiologia da PP. Exceto pela suspensão dos agentes causais e fotoproteção, não há tratamento baseado em evidência disponível até o momento, havendo relatos de controle clínico após o uso de N-acetilcisteína oral.

76
PSEUDOPORFIRIA A PP apresenta semelhança clínicohistopatológica da porfiria cutânea tardia, porém, pode ser distinguida pela normalidade dos níveis séricos, urinários e nas fezes de porfirinas. Ambas podem estar presentes nos pacientes renais crônicos Após a suspensão da droga, ainda podem surgir novas lesões por até 5 semanas. A fragilidade da pele persiste por 6 meses.

79
Obrigada !!!

Apresentações semelhantes













>")





, é transmitida de pessoa a pessoa, através da água e de alimentos contaminados com matéria fecal .EM.>")



