Carregar apresentação
A apresentação está carregando. Por favor, espere

1
ANEMIA FALCIFORME

2
Introdução Estrutura da Hemoglobina:
A hemoglobina é uma molécula de proteína composta de dois pares de cadeias globínicas, polipeptídicas. Cada cadeia contém uma molécula heme que é responsável pelo transporte de oxigênio. As cadeias globínicas são semelhantes umas as outras, consistindo de uma série de aminoácidos. A cadeia beta possui 146 aminoácidos e a cadeia alfa 141. Além das cadeias alfa (α ) e beta (β) que constituem a hemoglobina A (de adulto ), os tipos de cadeias polipeptídicas variam de acordo com o estádio de desenvolvimento intra-uterino. Todas são chamadas por letras gregas : gama (γ), delta (δ), épsilon (ε) e zeta (ζ). As cadeias zeta e épsilon são sintetizadas no início da vida intra-uterina ; as cadeias alfa e gama, na vida fetal e as cadeias alfa, beta e delta na vida pós fetal.
 e beta (β) que constituem a hemoglobina A (de adulto ), os tipos de cadeias polipeptídicas variam de acordo com o estádio de desenvolvimento intra-uterino. Todas são chamadas por letras gregas : gama (γ), delta (δ), épsilon (ε) e zeta (ζ). As cadeias zeta e épsilon são sintetizadas no início da vida intra-uterina ; as cadeias alfa e gama, na vida fetal e as cadeias alfa, beta e delta na vida pós fetal.")
3
Etiopatogenia A anemia falciforme é causada por uma mutação no gen da hemoglo-bina que produz uma anormalidade na cadeia beta globínica. A hemoglobina falciforme (Hb S) é um exemplo de mutação qualitativa na molécula da hemoglobina. Os estudos demostraram que na cadeia β na posição do Carbono 6 houve uma substituição do ácido glutâmico pela valina . Essa mutação é a responsável pela tendência da HbS desoxigenada formar polímeros e tomar a forma afoiçada. A valina é um aminoácido neutro enquanto que o ácido glutâmico é carregado negativamente. Assim a valina permite a aproximação das moléculas de hemoglobina quando desoxigenada e consequentemente a sua polimerização. Os estudos em microscopia eletrônica tem mostrado que as células irreversivelmente afoiçadas praticamente não tem hemoglobinas polimerizadas e ainda, retém a forma afoiçada. Assim, a forma "em foice" das células é devida a uma fixação das proteínas do citoesqueleto da membrana que devem então serem responsabilizadas pela forma irreversivelmente afoiçada.
 é um exemplo de mutação qualitativa na molécula da hemoglobina. Os estudos demostraram que na cadeia β na posição do Carbono 6 houve uma substituição do ácido glutâmico pela valina . Essa mutação é a responsável pela tendência da HbS desoxigenada formar polímeros e tomar a forma afoiçada. A valina é um aminoácido neutro enquanto que o ácido glutâmico é carregado negativamente. Assim a valina permite a aproximação das moléculas de hemoglobina quando desoxigenada e consequentemente a sua polimerização. Os estudos em microscopia eletrônica tem mostrado que as células irreversivelmente afoiçadas praticamente não tem hemoglobinas polimerizadas e ainda, retém a forma afoiçada. Assim, a forma em foice das células é devida a uma fixação das proteínas do citoesqueleto da membrana que devem então serem responsabilizadas pela forma irreversivelmente afoiçada.")
4
Fisiopatologia Quanto maios a concentração de hemoglobina siclêmica nas hemácias maior a probabilidade de ocorrer a polimerização da hemoglobina. O principal fator desencadeador de polimerização é a desoxigenação. Deve-se considerar também que uma elevação da Concentração da Hemoglobina Corpuscular Média (CHCM) resulta da desidratação das hemácias e formam hemácias densas, com efeito deletério no processo de afoiçamento. A maior concentração de HbF dificulta a polimerização da hemoglobina. Por não ser “polimerizável” a HbF mantém as cadeias de HbS separadas. Para bem explicar o fenômeno do afoiçamento, os eventos ocorrem em uma seqüência: Inicialmente há polimerização da hemoglobina S, Segue-se a diminuição da flexibilidade das hemácias, Há a oclusão da microcirculação, Como conseqüência há hipoxia tissular, Ocorre lesão tissular que dispara o estímulo doloroso.
 resulta da desidratação das hemácias e formam hemácias densas, com efeito deletério no processo de afoiçamento. A maior concentração de HbF dificulta a polimerização da hemoglobina. Por não ser polimerizável a HbF mantém as cadeias de HbS separadas. Para bem explicar o fenômeno do afoiçamento, os eventos ocorrem em uma seqüência: Inicialmente há polimerização da hemoglobina S, Segue-se a diminuição da flexibilidade das hemácias, Há a oclusão da microcirculação, Como conseqüência há hipoxia tissular, Ocorre lesão tissular que dispara o estímulo doloroso.")
5
História O mapa de origem da mutação genética que produziu a anemia falciforme coincide com a região geográfica da África que tem altos índices de malária. Essas regiões constituem cinturões onde a malária é endêmica. Os negros que aí vivem teriam maior chance de sobrevivência se tivessem o "traço" AS do queaqueles com hemoglobina normal (AA). Existem 3 (três) tipos de anemia falciforme: a Banto, considerada muito grave; a Benin é grave; e a Senegal que é leve. O trânsito de populações no globo, com o tráfico de escravizados e a imigração européia para as Américas fez com que as hemoglobinopatias não se restringissem aos povos de origem. No Brasil, a anemia falciforme do tipo banto é prevalente em relação aos outros tipos. Acontece que a anemia falciforme não está limitada às pessoas classificadas comonegras no Brasil, mas encontra maior freqüência neste grupo. Pessoas auto-classificadas como brancas podem ter herdado os genes e ter a anemia.
. Existem 3 (três) tipos de anemia falciforme: a Banto, considerada muito grave; a Benin é grave; e a Senegal que é leve. O trânsito de populações no globo, com o tráfico de escravizados e a imigração. européia para as Américas fez com que as hemoglobinopatias não se restringissem aos povos de origem. No Brasil, a anemia falciforme do tipo banto é prevalente em relação aos outros tipos. Acontece que a anemia falciforme não está limitada às pessoas classificadas comonegras no Brasil, mas encontra maior freqüência neste grupo. Pessoas auto-classificadas como brancas podem ter herdado os genes e ter a anemia.")
6
Epidemiologia Estima-se que aproximadamente 7% da população mundial seja acometida pelos transtornos das hemoglobinas, representados, na sua maioria, pelas talassemias e pela doença falciforme. No caso da HbS, a maior prevalência ocorre na África tropical e entre os negros de países que participaram do tráfico de escravos. No Brasil, cerca de 0,1% a 0,3% da população negra é afetada pela doença e estima-se a existência de pelo menos dois milhões de portadores da HbS (heterozigotos). Na região Sudeste,a prevalência estimada de heterozigotos é de 2% na população geral e, entre os negros, de 6% a 10%. No Brasil, Alves observou que 78,6% dos óbitos devidos à doença falciforme ocorreram até os 29 anos de idade, e 37,5% concentraram-se nos menores de nove anos. A elevada letalidade, que abrange especialmente jovens, reflete a gravidade da doença. O reconhecimento tardio de tais doenças pode levar à morte nos primeiros anos de vida.
. Na região Sudeste,a prevalência estimada de heterozigotos é de 2% na população geral e, entre os negros, de 6% a 10%. No Brasil, Alves observou que 78,6% dos óbitos devidos à doença falciforme ocorreram até os 29 anos de idade, e 37,5% concentraram-se nos menores de nove anos. A elevada letalidade, que abrange especialmente jovens, reflete a gravidade da doença. O reconhecimento tardio de tais doenças pode levar à morte nos primeiros anos de vida.")
7
Outras formas mistas: S –D; S – E; S – lepore; S – quebec - chori
Avaliação de Tipos e Percentuais de Hemoglobina em: INDIVÍDUO NORMAL TRAÇO FALCÊMICO ANEMIA FALCIFORME HbA (α2β2) - 97% HbA (α2β2) - 57% HbA (α2β2) - 0% HbA2 (α2δ2) - 2% HbA2 (α2δ2) - 2% HbA2 (α2δ2) - 2% HbS (α2βs2) - 0% HbS (α2βs2) - 40% HbS (α2βs2) - 93% HbF (α2y2) - 1% HbF (α2γ2) - 1% HbF (α2γ2)Q) - 5% FORMAS DA DOENÇA FALCIFORME Hb A1 HbA2 Hb F Hb C Hb S SS < 3.5 2 – 20% 80 – 95% SC - 1 – 5% 45 – 50% S beta 3.5 – 7% 2 – 15% 80 – 92% S beta+o 5 – 30% % 2 – 10% 65 – 90% AS – traço 50 – 60% <3.5% <2% 35 – 45% Normal 95 – 98% Outras formas mistas: S –D; S – E; S – lepore; S – quebec - chori
 - 97% HbA (α2β2) - 57% HbA (α2β2) - 0% HbA2 (α2δ2) - 2% HbA2 (α2δ2) - 2% HbA2 (α2δ2) - 2% HbS (α2βs2) - 0% HbS (α2βs2) - 40% HbS (α2βs2) - 93% HbF (α2y2) - 1% HbF (α2γ2) - 1% HbF (α2γ2)Q) - 5% FORMAS DA DOENÇA FALCIFORME. Hb A1. HbA2. Hb F. Hb C. Hb S. SS. < – 20% 80 – 95% SC. - 1 – 5% 45 – 50% S beta. 3.5 – 7% 2 – 15% 80 – 92% S beta+o. 5 – 30% % 2 – 10% 65 – 90% AS – traço. 50 – 60% <3.5% <2% 35 – 45% Normal. 95 – 98% Outras formas mistas: S –D; S – E; S – lepore; S – quebec - chori.")
8
DIAGNÓSTICO LABORATORIAL
Hemograma Anemia moderada Hb (6 – 10g/dl), Ht (18 – 30%), tende a ser N/N. Presença de hemácias afoiçadas (drepanócitos) – pode existir no AS. Inclusões nas hemácias (Howell-Jolly etc) – hipoesplenismo. Hemácias em alvo, eritroblastose, ponteado basofílico. Leucocitose com neutrofilia ( – /mm3 ) Trombocitose – Vários estímulos e hipoesplenismo VSH – Hemácia em foice não sedimentem bem. Bioquímica – (típica da hemólise). Elevação da bilirrubina indireta. (Se elevar a direta também, pensar em litíase biliar). Hptoglobina baixa. Elevação da DHL. Teste de falcização – Precisão limitada. Falsos negativos. Eletroforese de hemoglobina – Detecção da HbS e quantificação de cada tipo. PCR – Método usado no diagnóstico pré-natal.
, Ht (18 – 30%), tende a ser N/N. Presença de hemácias afoiçadas (drepanócitos) – pode existir no AS. Inclusões nas hemácias (Howell-Jolly etc) – hipoesplenismo. Hemácias em alvo, eritroblastose, ponteado basofílico. Leucocitose com neutrofilia ( – /mm3 ) Trombocitose – Vários estímulos e hipoesplenismo. VSH – Hemácia em foice não sedimentem bem. Bioquímica – (típica da hemólise). Elevação da bilirrubina indireta. (Se elevar a direta também, pensar em litíase biliar). Hptoglobina baixa. Elevação da DHL. Teste de falcização – Precisão limitada. Falsos negativos. Eletroforese de hemoglobina – Detecção da HbS e quantificação de cada tipo. PCR – Método usado no diagnóstico pré-natal.")
9
Clínica da Anemia Falciforme
INFECÇÃO Infecção Infantil Febre em criança com menos de 5 anos de idade geralmente indica infecção bacteriana grave. Há grande possibilidade da infecção por meningite pneumocócica ou infecção por H.influenzae. Infecção em Adultos As infecções tendem a ocorrer em áreas com lesões como: pulmões, rins e ossos. As infecções que ocorrem após procedimentos cirúrgicos são frequentes, principalmente após cirurgias de cabeça de fêmur ou colecistectomia. Infecção do trato urinário. Osteomielite: é mandatório estabelecer-se o diagnóstico bacteriano antes de se iniciar o tratamento. É importante realizar-se pesquisa dos microorganismos : Salmonella, Estafilococus e M. Tuberculosis.

10
Clínica da Anemia Falciforme
Crise de Hipersequestro Esplênico (CHSE) Ocorre principalmente em crianças. O pico de incidência vai dos 5 meses aos 2 anos. Após esta faixa os episódios são raros. Em alguns casos pode vir associada a infecçäo viral. Durante as crises graves de seqüestro, o baço torna-se volumoso, ocupando todo o abdome, às vezes indo até a região pélvica. A hemoglobina cai rapidamente (1 a 2 g/dl) podendo ocorrer choque hipovolêmico. A criança pode evoluir para o óbito poucas horas após os primeiros sinais desta complicação. As indicações clínicas da CHSE são astenia súbita, intensa palidez cutâneo mucosa, taquipnéia, taquicardia, aumento do volume abdominal e vertigens. A contagem de reticulocitos tende a estar aumentada. O tratamento das crises graves de hipersequestro esplênico é dirigido à pronta correção da hipovolemia com expansores plasmáticos e hemotransfusões. Os pacientes que apresentem mais de uma crise de hipersequestro devem ser encaminhados a esplenectonomia, devendo receber vacinas anti pneumococos e anti influenzae, além de penicilinoterapia profilática.
 Ocorre principalmente em crianças. O pico de incidência vai dos 5 meses aos. 2 anos. Após esta faixa os episódios são raros. Em alguns casos pode vir associada a infecçäo viral. Durante as crises graves de seqüestro, o baço torna-se volumoso, ocupando todo o abdome, às vezes indo até a região pélvica. A hemoglobina cai rapidamente (1 a 2 g/dl) podendo ocorrer choque hipovolêmico. A criança pode evoluir para o óbito poucas horas após os primeiros sinais desta complicação. As indicações clínicas da CHSE são astenia súbita, intensa palidez cutâneo mucosa, taquipnéia, taquicardia, aumento do volume abdominal e vertigens. A contagem de reticulocitos tende a estar aumentada. O tratamento das crises graves de hipersequestro esplênico é dirigido à pronta correção da hipovolemia com expansores plasmáticos e hemotransfusões. Os pacientes que apresentem mais de uma crise de hipersequestro devem ser encaminhados a esplenectonomia, devendo receber vacinas anti pneumococos e anti influenzae, além de penicilinoterapia profilática.")
11
Clínica da Anemia Falciforme
CRISES APLÁSTICAS Os pacientes apresentam-se com astenia, palidez cutâneo mucosa intensa e reticulocitopenia. Geralmente ocorre após infecção por parvovírus. A anemia é acentuada durante este período. A melhora ocorre em 5 a 10 dias. As amostras de sangue devem ser obtidas para a pesquisa de parvovirose (IgG e IgM). O tratamento é sintomático com transfusões. Deve ser avisado à família para que as demais crianças com Doença Falciforme do grupo familiar sejam observadas pois podem estar contaminadas e apresentarem os mesmos sintomas (proceder ao estudo sorológico da parvovirose no grupo familiar próximo ao paciente em questão).
. O tratamento é sintomático com transfusões. Deve ser avisado à família para que as demais crianças com Doença. Falciforme do grupo familiar sejam observadas pois podem estar. contaminadas e apresentarem os mesmos sintomas (proceder ao estudo sorológico da parvovirose no grupo familiar próximo ao paciente em questão).")
12
Clínica da Anemia Falciforme
CRISES ALGICAS Geralmente são devidas à lesões isquêmicas secundárias à obstrução do fluxo sangüíneo pelas hemácias afoiçadas. O evento doloroso geralmente dura de 4 a 6 dias, às vezes persistindo por semanas. Eventos precipitantes: hipoxia, infecção, acidose, desidratação, resfriamento da pele,gestação, ansiedade e depressão. Tratamento Identificação do(s) fator(es) precipitante(s). Hidratação: correção dos déficits de líquidos e eletrólitos e continuação da administração de volume uma vez corrigidas as deficiências. Analgésicos: deve-se iniciar com medicamentos que não sejam narcóticos. Raramente serão necessários narcóticos mais potentes como a Meperidina (1,5 mg/Kg/dose cada 2-4 horas) IM ou IV. Prevenção da dor: evitar hipoxia, desidratação, resfriamento da pele, natação em água fria. Em casos de crise álgidas de repetição anotar o dia do início e do final da crise para ser estudado o intervalo destas. A hidroxiuréia, com eficácia comprovada nas crises dolorosas e STA pelo aumento da concentração de Hb F, pode reduzir a frequência dos episódios de priapismo, no entanto faltam estudos randomizados para comprovar seu benefício para esta complicação.
 fator(es) precipitante(s). Hidratação: correção dos déficits de líquidos e eletrólitos e continuação da administração de volume uma vez corrigidas as deficiências. Analgésicos: deve-se iniciar com medicamentos que não sejam narcóticos. Raramente serão necessários narcóticos mais potentes como a Meperidina (1,5 mg/Kg/dose cada 2-4 horas) IM ou IV. Prevenção da dor: evitar hipoxia, desidratação, resfriamento da pele, natação em água fria. Em casos de crise álgidas de repetição anotar o dia do início e do final da crise para ser estudado o intervalo destas. A hidroxiuréia, com eficácia comprovada nas crises dolorosas e STA pelo aumento da concentração de Hb F, pode reduzir a frequência dos episódios de priapismo, no entanto faltam estudos randomizados para comprovar seu benefício para esta complicação.")
13
SÍNDROME TORÁCICA AGUDA
Clínica da Anemia Falciforme SÍNDROME TORÁCICA AGUDA Qualquer infiltrado novo acometendo pelo menos um segmento de um lobo pulmonar, independente da etiologia infecciosa ou não, considerar como Síndrome Torácica Aguda (STA). Segundo alguns autores a mortalidade é de cerca de 25%. É sabido que 50% de todos pacientes com Doença Falciforme tiveram pelo menos 1 episódio de STA. Etiologia Infecciosa: Pneumococcus, E.Coli, H.Influenzae, Klebsiella, Chlamydia pneumoniae, Mycoplasma, Legionella, Vírus - Influenzae, RSV, CMV, Parvovírus, Adenovírus Parainfluenzae. Não infecciosa: infarto pulmonar, atelectasia pulmonar, edema pulmonar, trombose de veia profunda, infarto de costela.
. Segundo alguns autores a mortalidade é de cerca de 25%. É sabido que 50% de todos pacientes com Doença Falciforme tiveram pelo menos 1 episódio de STA. Etiologia. Infecciosa: Pneumococcus, E.Coli, H.Influenzae, Klebsiella, Chlamydia pneumoniae, Mycoplasma, Legionella, Vírus - Influenzae, RSV, CMV, Parvovírus, Adenovírus Parainfluenzae. Não infecciosa: infarto pulmonar, atelectasia pulmonar, edema pulmonar, trombose de veia profunda, infarto de costela.")
14
Clínica da Anemia Falciforme
Diagnóstico Clínico da STA. A STA pode se desenvolver como um evento isolado ou durante o curso de uma crise álgica vásculo-oclusiva. A dor pleurítica é o sintoma predominante em adultos. Nas crianças pequenas, a febre, a tosse e a taquipnéia são geralmente os únicos sinais e sintomas . O acometimento da pleura diafragmática pode levar à dor abdominal. Ao exame físico , geralmente nota-se taquipnéia e, algumas vezes ,atrito pulmonar. Podem também ser evidenciados sinais de consolidação pulmonar e derrame pleural. Diagnóstico Laboratorial Ao RX de tórax - infiltrado em um ou mais lobos com ou sem derrame pleural. Proceder-se-á à bacterioscopia e cultura de escarro, hemocultura, estudo do líquido pleural, assim como a lavagem brônquica para a pesquisa de bactérias e vírus. Deverá ser verificada a gasometria arterial ou oximetria de pulso. Tratamento Analgésicos / evitar narcóticos Hidratação e Oxigenoterapia A maioria dos pacientes deve ser tratada com Penicilina, Ampicilina ou Cefalosporina. A eritracitaferese (doença de progressão grave

15
Clínica da Anemia Falciforme
SÍNDROME DA EMBOLIA GORDUROSA Esta complicação é rara, mas geralmente fatal. É devida à embolização generalizada de gordura da medula óssea para vasos pulmonares e daí para circulação sistêmica. ACIDENTE VASCULAR CEREBRAL (AVC) No AVC há uma constelação de sinais neurológicos devido a uma lesão isquêmica ou Hemorrágica num território vascular específico. O AVC ocorre em 6 a 12% dos pacientes. GRAVIDEZ E ANEMIA FALCIFORME A gravidez acarreta um aumento da morbidade e mortalidade para a mulher com Anemia Falciforme e para o seu concepto. Porém os riscos não são grandes o suficiente para se proibir as gestações desejadas. COLELITÍASE E COLEDOCOLITÍASE A bilirrubina sérica está aumentada nos pacientes com An Falciforme devido a hemólise sangüínea. Quando o fígado está funcionando normalmente, a bilirrubina sérica total não deve exceder 4mg/dl e a fração conjugada (direta), 10% do total. Com o passar do tempo a alta taxa de excreção de bilirrubinas tem como conseqüência a formação de cálculos biliares. São encontrados em aproximadamente 14% das crianças menores de 10 anos, cerca de 30% em adolescentes e 75 % em adultos com 30 anos.
 No AVC há uma constelação de sinais neurológicos devido a uma lesão isquêmica ou. Hemorrágica num território vascular específico. O AVC ocorre em 6 a 12% dos pacientes. GRAVIDEZ E ANEMIA FALCIFORME. A gravidez acarreta um aumento da morbidade e mortalidade para a mulher com Anemia. Falciforme e para o seu concepto. Porém os riscos não são grandes o suficiente para se. proibir as gestações desejadas. COLELITÍASE E COLEDOCOLITÍASE. A bilirrubina sérica está aumentada nos pacientes com An Falciforme devido a hemólise. sangüínea. Quando o fígado está funcionando normalmente, a bilirrubina sérica total não deve exceder. 4mg/dl e a fração conjugada (direta), 10% do total. Com o passar do tempo a alta taxa de excreção de bilirrubinas tem como conseqüência a. formação de cálculos biliares. São encontrados em aproximadamente 14% das crianças menores de 10 anos, cerca de 30% em adolescentes e 75 % em adultos com 30 anos.")
16
Clínica da Anemia Falciforme
OSSOS E ARTICULAÇÕES As lesões podem resultar de hiperplasia de medula óssea como na gnatopatia ou de infarto ósseo. Exemplos de infarto são a síndrome mão-pé em crianças. Infartos ósseos múltiplos em crianças mais velhas e adultos e necrose avascular da cabeça do fêmur e de outros ossos. Necrose Avascular O paciente queixa-se de dor persistente na região inguinal ou nádegas. A cirurgia com prótese pode ser feita na fase inicial da necrose avascular afim de se tentar parar o processo necrótico. Atualmente o tratamento consiste em repouso articular, calor local e analgésicos. Síndrome mão-pé É quase que exclusivamente da criança pequena. Há dor, febre baixa e edema difuso, sem cacifo no dorso das mãos e dos pés que se estende aos dedos. O tratamento é feito com repouso, hidratação e analgesia. Outros Ossos e Síndromes Articulares Possíveis etiologias: infarto ósseo, edema inflamatório, gota, infarto sinovial, artrite séptica ou concomitância de doença reumática ou doença do colágeno com osteoartrite.

17
Clínica da Anemia Falciforme
PRIASPISMO O priapismo é uma ereção persistente e dolorosa do pênis. A fisiopatologia do priapismo ainda não está bem esclarecida. O fluxo venoso do corpo cavernoso nem sempre está totalmente ocluído como antes havia sido postulado. Os objetivos do tratamento são: esvaziar o corpo cavernosos; melhorar a dor do paciente; prevenir a impotência. Sem intervenções, o priapismo severo provavelmente leva à perda parcial ou completa da função erétil em tono de 80% dos casos. Em melhores circunstâncias, as transfusões e cirurgia diminuem esta incidência em % dos casos, se bem realizada. O dietil-estilbestrol é uma droga anti-androgênica que, apesar dos poucos relatos na literatura, vem se mostrando eficaz no tratamento do priapismo, reduzindo sua frequência e duração.

18
Clínica da Anemia Falciforme
1. Recomendações gerais Repouso com o membro elevado Analgesia (usar opióides se necessário) 2. Tratamento local Debridamento com compressas molhadas (em contato com a úlcera) a secas (parte externa do curativo) Pomadas Zinco tópico Antibióticos tópicos Bota de Unna Hidroterapia Câmara hiperbárica 3. Tratamento cirúrgico Debridamento cirúrgico Enxerto de pele 4. Tratamento sistêmico Transfusões sanguíneas Antibióticos sistêmicos Sulfato de zinco oral Hidroxiuréia As úlceras de perna Ocorrem uni ou bilateralmente, principalmente nos maléolos laterais e mediais dos tornozelos, mas podem se desenvolver no dorso dos pése nas pernas, e existem raros relatos desta complicação nas mãos. Como as extremidades inferiores do corpo são locais expostos, traumas locais e picadas de inseto podem precipitar a formação das úlceras (figura 6.39). O exame físico das extremidades é essencial para detectar alterações na pele do paciente e úlceras em fase inicial. A prevalência das úlceras de membros inferiores é por volta de 2,5% entre os doentes, sendo mais comum nos pacientes com HbSS (cerca de 5%), e raras naqueles com HbSC e Sb -talassemia. Praticamente não ocorre antes dos 10 anos de idade. A incidência é maior após os 20 anos, principalmente no sexo masculino, nos pacientes com anemia falciforme (10 casos por 100 pacientes/ano).
 2. Tratamento local. Debridamento com compressas molhadas (em contato com a úlcera) a secas (parte externa do curativo) Pomadas. Zinco tópico. Antibióticos tópicos. Bota de Unna. Hidroterapia. Câmara hiperbárica. 3. Tratamento cirúrgico. Debridamento cirúrgico. Enxerto de pele. 4. Tratamento sistêmico. Transfusões sanguíneas. Antibióticos sistêmicos. Sulfato de zinco oral. Hidroxiuréia. As úlceras de perna. Ocorrem uni ou bilateralmente, principalmente. nos maléolos laterais e mediais dos tornozelos, mas podem se desenvolver no dorso dos pése. nas pernas, e existem raros relatos desta. complicação nas mãos. Como as extremidades. inferiores do corpo são locais expostos, traumas. locais e picadas de inseto podem precipitar a. formação das úlceras (figura 6.39). O exame. físico das extremidades é essencial para detectar. alterações na pele do paciente e úlceras em. fase inicial. A prevalência das úlceras de membros inferiores. é por volta de 2,5% entre os doentes, sendo mais. comum nos pacientes com HbSS (cerca de 5%), e raras naqueles com HbSC e Sb -talassemia. Praticamente não ocorre antes dos 10 anos de. idade. A incidência é maior após os 20 anos, principalmente no sexo masculino, nos pacientes. com anemia falciforme (10 casos por 100. pacientes/ano).")
19
MÉTODOS TRANSFUSIONAIS
Transfusões simples:Podem ser empregadas para anemia aguda ou hipovolêmica ou num programa de transfusões a longo prazo. 2) Eritracitaferese: É empregada quando se deseja aumentar rapidamente o nível de hemoglobina com substituição das hemácias afoiçadas por hemácias normais. COMPLICAÇÕES DAS TRANSFUSÕES SOBRECARGA DE VOLUME: Ocorre quando muito volume é transfundido rapidamente principalmente em pacientes com insuficiência cardíaca. SOBRECARGA DE FERRO: Todos os pacientes hipertransfundidos devem ter a ferritina sérica medida periodicamente. ALOIMUNIZAÇÃO A incidência de aloimunização nos pacientes com Doença Falciforme é em torno de 20-25%. A redução de reações transfusionais hemolíticas e aloimunização pode ser conseguida seguindo-se as recomendações; Manter registradas todas as transfusões e suas complicações; Limitar o número de transfusões administradas; Pesquisar anticorpos cada 1 a 2 meses pós transfusão; Diminuir a oportunidade de aloimunizações, fenotipando o sangue do paciente e do doador;
 Eritracitaferese: É empregada quando se deseja aumentar rapidamente o nível de hemoglobina com substituição das hemácias afoiçadas por hemácias normais. COMPLICAÇÕES DAS TRANSFUSÕES. SOBRECARGA DE VOLUME: Ocorre quando muito volume é transfundido rapidamente principalmente em pacientes com insuficiência cardíaca. SOBRECARGA DE FERRO: Todos os pacientes hipertransfundidos devem ter a ferritina sérica medida periodicamente. ALOIMUNIZAÇÃO. A incidência de aloimunização nos pacientes com Doença Falciforme é em torno de 20-25%. A redução de reações transfusionais hemolíticas e aloimunização pode ser conseguida seguindo-se as recomendações; Manter registradas todas as transfusões e suas complicações; Limitar o número de transfusões administradas; Pesquisar anticorpos cada 1 a 2 meses pós transfusão; Diminuir a oportunidade de aloimunizações, fenotipando o sangue do paciente e do doador;")
20
TALASSEMIAS

21
Definição As talassemias são um grupo heterogêneo de doenças genéticas causadas pela redução da síntese de globinas alfa e não-alfa (b, d ou g). Na realidade as formas mais comuns de talassemias se devem à redução de globina alfa ou de globina beta, situações que originam as talassemias alfa ou beta, respectivamente. Situações mais raras envolvem a redução de síntese conjunta de globinas delta e beta (talassemia d b), ou de delta, beta e gama (talassemia d b g). Em alguns casos de talassemias há redução total de síntese de globina alfa ou de beta, caracterizando as talassemias alfa0 ou beta 0, respectivamente, por outro lado quando a redução de síntese é parcial denomina-se por talassemias a+ ou b +. Pelo fato da talassemia beta, bem como as hemoglobinas S, C e E serem as mais prevalentes respectivamente nos continentes europeu, africano e asiático, não é raro a ocorrência de interações, combinações entre genes talassêmicos com Hb S, principalmente, produzem grande diversidade clínica dessa doença genética, com variações que causam desde a morte fetal intra-útero até situações assintomáticas.
, ou de delta, beta e gama (talassemia d b g). Em alguns casos de talassemias há redução total de síntese de globina alfa ou de beta, caracterizando as talassemias alfa0 ou beta 0, respectivamente, por outro lado quando a redução de síntese é parcial denomina-se por talassemias a+ ou b +. Pelo fato da talassemia beta, bem como as hemoglobinas S, C e E serem as mais prevalentes respectivamente nos continentes europeu, africano e asiático, não é raro a ocorrência de interações, combinações entre genes talassêmicos com Hb S, principalmente, produzem grande diversidade clínica dessa doença genética, com variações que causam desde a morte fetal intra-útero até situações assintomáticas.")
22
Epidemiologia e generalidades
É importante destacar que as talassemias alfa podem ter duas causas de origem hereditária e adquirida. Evidentemente as formas hereditárias são as mais comuns e atingem, pelo menos, 20 dos brasileiros dos quais 17% são assintomáticos e com valores hematimétricos (Hb, Ht, VCM e HCM) normais, e 3% tem discretos graus de anemia microcítica e hipocrômica. 1:5.000 pessoas é portadora da doença de Hb H). As formas adquiridas são secundárias a um processo patológico primário, por exemplo doenças linfo e mieloproliferativas, anemia sideroblástica, etc.
 normais, e 3% tem discretos graus de anemia microcítica e hipocrômica. 1:5.000 pessoas é portadora da doença de Hb H). As formas adquiridas são secundárias a um processo patológico primário, por exemplo. doenças linfo e mieloproliferativas, anemia sideroblástica, etc.")
24
Esquema 1 - Tipos de lesões que causam talassemia alfa.
Classificação As talassemias alfa são diferenciadas e classificadas de acordo com o número de genes alfa lesados, com a importante observação de que o grau de lesão pode ser variável, afetando o gene parcial ou totalmente. De uma forma geral, representa-se uma pessoa sem talassemia alfa com seus quatro genes alfa funcionantes (a ,a /a ,a ), sendo dois genes alfa de um cromossomo 16 e dois do outro cromossomo 16, provenientes um do pai e outro da mãe. Esquema 1 - Tipos de lesões que causam talassemia alfa.
, sendo dois genes alfa de um cromossomo 16 e dois do outro cromossomo 16, provenientes um do pai e outro da mãe. Esquema 1 - Tipos de lesões que causam talassemia alfa.")
25
Tipos de αTalassemia Portador “silencioso” – é o tipo mais comum entre as talassemias alfa e se deve à deleção de apenas um gene alfa (-, a /a ,a ). O portador desse tipo de talassemia é assintomático, e embora o volume corpuscular médio (VCM) se apresente como discretamente microcítico (VCM < 80), Traço alfa talassêmico – se deve à deleção de dois genes alfa (-,-/a ,a ) ou (-,a /-,a ). Os portadores, apesar de serem normais sob o ponto de vista clínico, reclamam de fraqueza, cansaço, dores nas pernas e palidez. Doença de Hb H – é causada pela deleção de três genes alfa (-,-/-,a ) . Essa patologia se expressa com uma forma moderadamente grave de talassemia, caracterizada por anemia microcítica e hipocrômica, hemoglobina total variável entre 8 e 11g/dL, aumento do baço e do fígado. Hidropsia Fetal – É a forma mais grave de todos os tipos de talassemias (alfa e beta), pois é uma forma letal. É uma situação comum no Extremo Asiático, sendo, entretanto, esporádica no Brasil. Os recém-nascidos afetados pela deleção dos quatro genes alfa (-,-/-,-) apresentam anemia muito grave, com hemoglobina inferior a 7g/dL, eritroblastose fetal, edema, grande aumento do baço e do fígado, e morte com poucas horas após o nascimento. Eletroforeticamente, a concentração de Hb Bart’s, está entre 80 e 100%, e a Hb H entre 10 e 20%.
. O portador desse tipo de talassemia é assintomático, e embora o volume corpuscular médio (VCM) se apresente como discretamente microcítico (VCM < 80), Traço alfa talassêmico – se deve à deleção de dois genes alfa (-,-/a ,a ) ou (-,a /-,a ). Os portadores, apesar de serem normais sob o ponto de vista clínico, reclamam de fraqueza, cansaço, dores nas pernas e palidez. Doença de Hb H – é causada pela deleção de três genes alfa (-,-/-,a ) . Essa patologia se expressa com uma forma moderadamente grave de talassemia, caracterizada por anemia microcítica e hipocrômica, hemoglobina total variável entre 8 e 11g/dL, aumento do baço e do fígado. Hidropsia Fetal – É a forma mais grave de todos os tipos de talassemias (alfa e beta), pois é uma forma letal. É uma situação comum no Extremo Asiático, sendo, entretanto, esporádica no Brasil. Os recém-nascidos afetados pela deleção dos quatro genes alfa (-,-/-,-) apresentam anemia muito grave, com hemoglobina inferior a 7g/dL, eritroblastose fetal, edema, grande aumento do baço e do fígado, e morte com poucas horas após o nascimento. Eletroforeticamente, a concentração de Hb Bart’s, está entre 80 e 100%, e a Hb H entre 10 e 20%.")
26
Beta Talassemias - Introdução
As talassemias beta são mais heterogêneas do que as do tipo alfa. Caracterizam-se por uma alteração quantitativa da síntese de globinas beta. São classificadas como talassemias beta zero (ou talassemia b 0) quando não há síntese de globinas, e talassemias beta mais (ou talassemia b+) quando há alguma taxa de síntese. Consequentemente as globinas alfa, que são sintetizadas normalmente, acumulam-se nos eritrócitos, durante a eritropoiese, causando agregação e precipitação. Os precipitados, formados em quantidades variáveis, danificam a membrana e destroem prematuramente essas células provocando a anemia. Com a utilização de técnicas de biologia molecular foi possível a identificação de aproximadamente 180 tipos diferentes de talassemias beta, cujas diversidades estão relacionadas com os graus de lesões no gene beta, podendo inclusive atingir os genes delta, pseudogene beta-1, os genes gama alanina e gama glicina e até o gene embrionário épsilon.
 quando não há síntese de globinas, e talassemias beta mais (ou talassemia b+) quando há alguma taxa de síntese. Consequentemente as globinas alfa, que são sintetizadas normalmente, acumulam-se nos eritrócitos, durante a eritropoiese, causando agregação e precipitação. Os precipitados, formados em quantidades variáveis, danificam a membrana e destroem prematuramente essas células provocando a anemia. Com a utilização de técnicas de biologia molecular foi possível a identificação de aproximadamente 180 tipos diferentes de talassemias beta, cujas diversidades estão relacionadas com os graus de lesões no gene beta, podendo inclusive atingir os genes delta, pseudogene beta-1, os genes gama alanina e gama glicina e até o gene embrionário épsilon.")
27
Beta Talassemia Quadro clínico da beta talassemia
Na beta talassemia, o principal responsável pelo desenvolvimento da anemia é a eritropoiese ineficaz com hemólise intra-medular dos precursores eritróides. Assim, nas apresentações mais leves da doença, a eritropoiese ineficaz é de cerca de 25%, enquanto que nos casos mais graves pode chegar até 90%. Isso justifica a discrepância comumente observada entre o aumento relativamente modesto do número de reticulócitos na vigência de quadros anêmicos muito intensos associados à hiperplasia acentuada da medula óssea.

28
Beta Talassemia menor ou minor (Traço talassêmico)
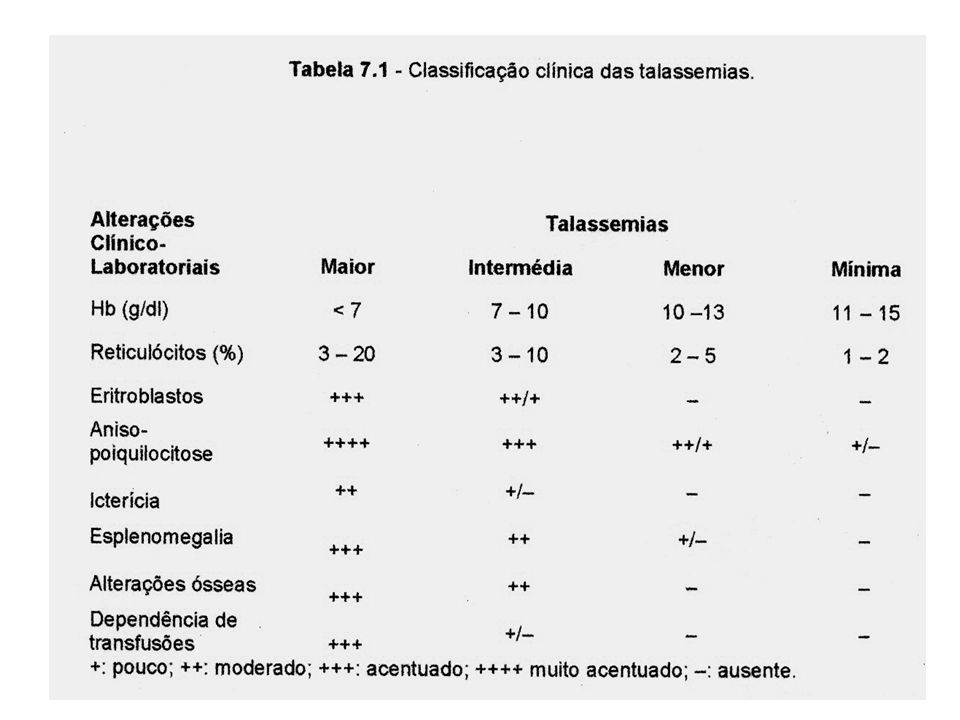
Resumo clínico: Herança: heterozigoto Valor basal da hemoglobina: ³9,5-10g/dL Sintomas: ausentes ou de leve intensidade. Transfusões:não são necessárias 2) Beta Talassemia intermediária (Intermédia) Resumo clínico: Herança: homozigoto ou heterozigoto Valor basal da hemoglobina: 7-9g/dL Sintomas: presentes e de moderada intensidade Transfusões: ocasionais 3) Beta Talassemia maior ou major Resumo clínico: Herança: homozigoto Valor basal da hemoglobina: <7g/dL Sintomas: presentes e de intensidade acentuada Transfusões: regulares Sobrecarga de ferro: inevitável
 Beta Talassemia intermediária (Intermédia) Resumo clínico: Herança: homozigoto ou heterozigoto Valor basal da hemoglobina: 7-9g/dL Sintomas: presentes e de moderada intensidade. Transfusões: ocasionais. 3) Beta Talassemia maior ou major. Resumo clínico: Herança: homozigoto Valor basal da hemoglobina: <7g/dL Sintomas: presentes e de intensidade acentuada Transfusões: regulares Sobrecarga de ferro: inevitável.")
29
Tratamento das Talassemias
Esplenectomia É uma medida auxiliar quando as complicações exedem os benefícios da presença do baço. Duas indicações principais: 1- Plaquetopenia 2- Consumo transfusional acima de 240 ml/Kg/ano para manter Hb de 10g/dl. Pode aumentar o nível médio do valor da Hemoglobina. Adiar até os 5 anos, se possível. Proceder vacinação (controvertido. Antibioticoterapia profilática (Penicilina Benzatina).
.")
30
Tratamento Transfusões de sangue
Transfusões de sangue regulares desde a infância consistem na melhor e principal forma de tratamento para pacientes portadores de talassemia maior. Os principais objetivos do tratamento transfusional são: corrigir a anemia, permitir o crescimento, desenvolvimento e realização de atividades normais, prevenir o aumento do baço e inibir a hiperplasia eritróide na medula óssea. Geralmente costuma-se transfundir o paciente para manter o valor de hemoglobina ente 9,5 e 10g/dL. Nessas condições, é possível permitir o crescimento e desenvolvimento adequado das crianças afetadas, além de inibir a hiperplasia da medula óssea e minimizar o acúmulo de ferro. O intervalo entre as transfusões geralmente varia de 2 a 4 semanas e a quantidade de concentrados de hemácias varia de 1 a 3 unidades por vez (ou 10 a 15ml por Kg de peso). Complicações do tratamento transfusional A principal complicação relacionada ao regime transfusional crônico é a sobrecarga de ferro. Outras complicações importantes associadas às transfusões de hemácias são: • Reações transfusionais agudas (febril hemolítica e não-hemolítica, e urticariforme, entre outras) •Transmissão de infecções (p.ex. hepatites virais e HIV) • Alo-imunização
. Complicações do tratamento transfusional. A principal complicação relacionada ao regime transfusional crônico é a sobrecarga de ferro. Outras complicações importantes associadas às transfusões de hemácias são: • Reações transfusionais agudas (febril hemolítica e não-hemolítica, e urticariforme, entre outras) •Transmissão de infecções (p.ex. hepatites virais e HIV) • Alo-imunização.")
31
Tratamento Sobrecarga de Ferro - Quelação do Ferro
Desferrioxamina (Desferal®): é o quelante mais antigo e tradicional, utilizado como monoterapia por décadas. Devido ao grande tamanho da molécula desta droga, a absorção intestinal torna-se impossibilitada. Assim, seu principal inconveniente é a via de administração, necessariamente subcutânea ou endovenosa, e o tempo prolongado de infusão (Figura 6). Deferiprone (L1): foi o primeiro quelante oral. Deferasirox (Exjade®): novo quelante oral com resultados promissores e posologia cômoda. Transplante de medula óssea Até o momento, já foram transplantados mais de 1600 pacientes com talassemia maior. Na grande maioria dos casos este tratamento tem sido realizado em pacientes com menos de 17 anos de idade, sendo que a idade ideal para a realização deste procedimento situa-se entre os 1 meses e 3 anos.
: é o quelante mais antigo e tradicional, utilizado como monoterapia por décadas. Devido ao grande tamanho da molécula desta droga, a absorção intestinal torna-se impossibilitada. Assim, seu principal inconveniente é a via de administração, necessariamente subcutânea ou endovenosa, e o tempo prolongado de infusão (Figura 6). Deferiprone (L1): foi o primeiro quelante oral. Deferasirox (Exjade®): novo quelante oral com resultados promissores e posologia cômoda. Transplante de medula óssea Até o momento, já foram transplantados mais de 1600 pacientes com talassemia maior. Na grande maioria dos casos este tratamento tem sido realizado em pacientes com menos de 17 anos de idade, sendo que a idade ideal para a realização deste procedimento situa-se entre os 1 meses e 3 anos.")
Apresentações semelhantes


, e que pode levar a incapacitação funcional.>")

 Reação mais comum durante a transfusão Motivo:>")

, é transmitida de pessoa a pessoa, através da água e de alimentos contaminados com matéria fecal .EM.>")







>")




