Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Teresa Neto Mariana Neves Neuza Ferreira
Hematologia 4º ano Teresa Neto Mariana Neves Neuza Ferreira

2
Talassemias Síntese deficiente de uma ou mais cadeias polipeptídicas da hemoglobina humana normal Anemia+ esplenomegalia+ deformidades ósseas

3
Recordando…. A1 que representa 97% do total
A hemoglobina é uma molécula complexa, formada por uma porção proteica – Globulina - e uma não proteica - heme. No organismo humano encontram-se 3 tipos de Hb: A1 que representa 97% do total A2 que corresponde a 3% da Hb do adulto F presente durante a vida intra-uterina A diferença entre estas encontra-se na estrutura da globina que se compõe de dois pares de cadeias proteicas: A1: α2 β2 A2: α2δ2 F: α2 γ2 Durante os primeiros 6 meses de vida a HB F é substituída pela Hb do adulto (Hb A1) com a formação de cadeias β ao invés de γ. Quando tal não é possível a produção de HB F prolonga-se na vida extra uterina.
 com a formação de cadeias β ao invés de γ. Quando tal não é possível a produção de HB F prolonga-se na vida extra uterina.")
4
Hemoglobina Subunidade α-like Subunidade β- like Tempo de expressão Gower 1 ζ ε Embrionária Gower 2 α Portland γ HbF (fetal) Fetal HbA2 δ Pós-natal HbA (adulto) β
 Fetal. HbA2. δ. Pós-natal. HbA (adulto) β.")
5
Fracção da total hemoglobina
Tipo de hemoglobina Fracção da total hemoglobina HbA ~92% HbA1a 0.75% HbA1b 1.5% HbA1c 3-6% HbA2 2.5% Total 100%

6
Talassemia α Genes da talassemia α: Sudeste asiático e populações originárias da costa oeste africana Genes que codificam a cadeia α: 2 em cada cr 16 Delecções no cr 16 levam à doença

7
Transmissão de talassemia α

8
Patofisiologia: Diminui síntese de cadeias α » diminui síntese Hb » hipocromia » eritrocitose com aumento da quantidade de tecido eritróide » hepatomegalia e deformidades ósseas ø cadeias α » formação de agregados γ4 ou β4 » precipitam no citoplasma » danificação das membranas celulares » destruição dos eritrócitos » anemia agregados γ4 ou β4: instáveis e com alta afinidade para oxigénio, oxidação fácil e com tendência a precipitar » anóxia e destruição celular Destruição celular » aumento da produção de GR » expansão das cavidades medulares por tecido eritróide » deformidades ósseas e fracturas

9
Genótipos possíveis: (αα;αα) : normal
(αα;α-) : talassemia silenciosa; portador assintomático (α-;α-) : α-talassemia-2; portador assintomático (--;αα) : α-talassemia-1; portador assintomático (--;α-) : doença de Hb H (--;--) : doença Hb de Bart
 : talassemia silenciosa; portador assintomático. (α-;α-) : α-talassemia-2; portador assintomático. (--;αα) : α-talassemia-1; portador assintomático. (--;α-) : doença de Hb H. (--;--) : doença Hb de Bart.")
10
(αα;α-) : portador silencioso
Apenas um gene α sofreu delecção Assintomático Ø anomalias clínicas ou hematológicas Apenas uma minoria apresenta MCV e MCH diminuídos Pais ou filhos de doentes com Hb H

11
(α-;α-) : portador assintomático (--;αα) : portador assintomático
Dois genes α sofreram delecção Ásia, Mediterrâneo e África Hemoglobina normal ou ligeiramente reduzida Microcitose (MCV: fl), hipocromia e anisopoiquilocitose assintomáticas Estudo de síntese de globinas nos reticulócitos do sangue periférico: 25% de redução de síntese de cadeias α relativamente às cadeias β Raras inclusões citoplasmáticas de Hg H (β4) Técnicas de mapeamento de DNA: delecção de 2 genes estruturais de α globolina Sem sintomas
, hipocromia e anisopoiquilocitose assintomáticas. Estudo de síntese de globinas nos reticulócitos do sangue periférico: 25% de redução de síntese de cadeias α relativamente às cadeias β. Raras inclusões citoplasmáticas de Hg H (β4) Técnicas de mapeamento de DNA: delecção de 2 genes estruturais de α globolina. Sem sintomas.")
12
(--;α-) : doença de Hb H Três genes α sofreram delecção
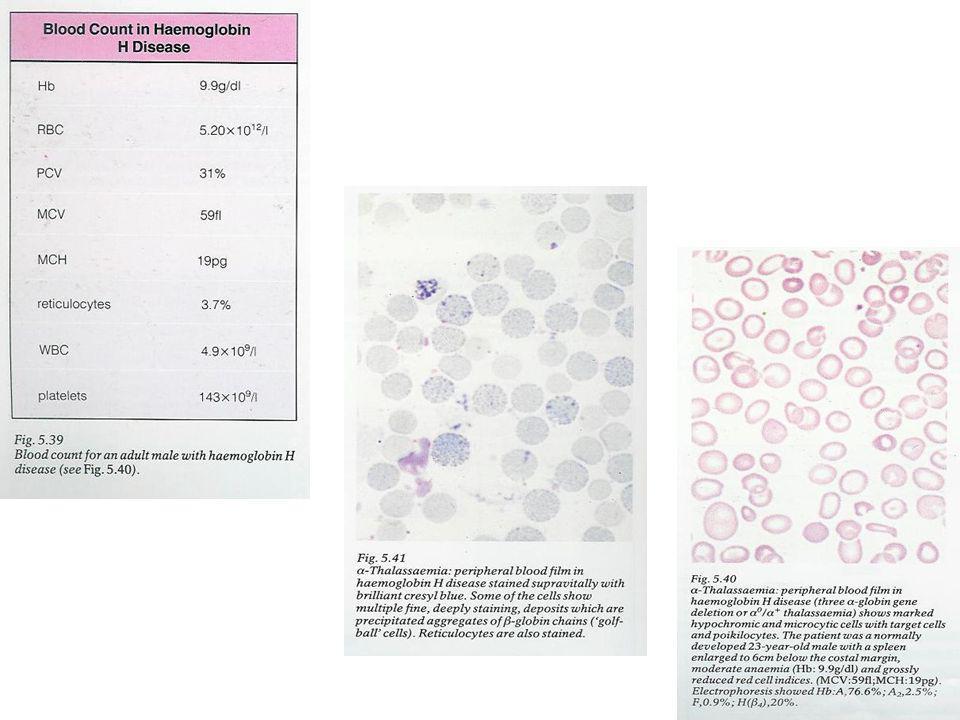
Sudeste asiático, ilhas mediterrânicas, Médio Oriente Redução da produção de cadeias α com formação de tetrâmeros β4 (Hb H): 5-30% Hb H é instável e precipita nos eritroblastos, mas não nos GR circulantes Crianças normais à nascença Anemia e esplenomegalia até 1 ano de idade Icterícia e hepatoesplenomegalia, possíveis anomalias ósseas e atraso mental Anemia hemolítica crónica moderada a severa Hb: g/dl (ou mais baixa); HbA1: 70-95% Reticulócitos: % Hipocromia, microcitose (MCV: fl) e células em alvo Electroforese: % Hb de Bart no nascimento » Hb H (5- 30 %)
: 5-30% Hb H é instável e precipita nos eritroblastos, mas não nos GR circulantes. Crianças normais à nascença. Anemia e esplenomegalia até 1 ano de idade. Icterícia e hepatoesplenomegalia, possíveis anomalias ósseas e atraso mental. Anemia hemolítica crónica moderada a severa. Hb: g/dl (ou mais baixa); HbA1: 70-95% Reticulócitos: % Hipocromia, microcitose (MCV: fl) e células em alvo. Electroforese: % Hb de Bart no nascimento » Hb H (5- 30 %)")
14
(--;--) : doença Hb de Bart
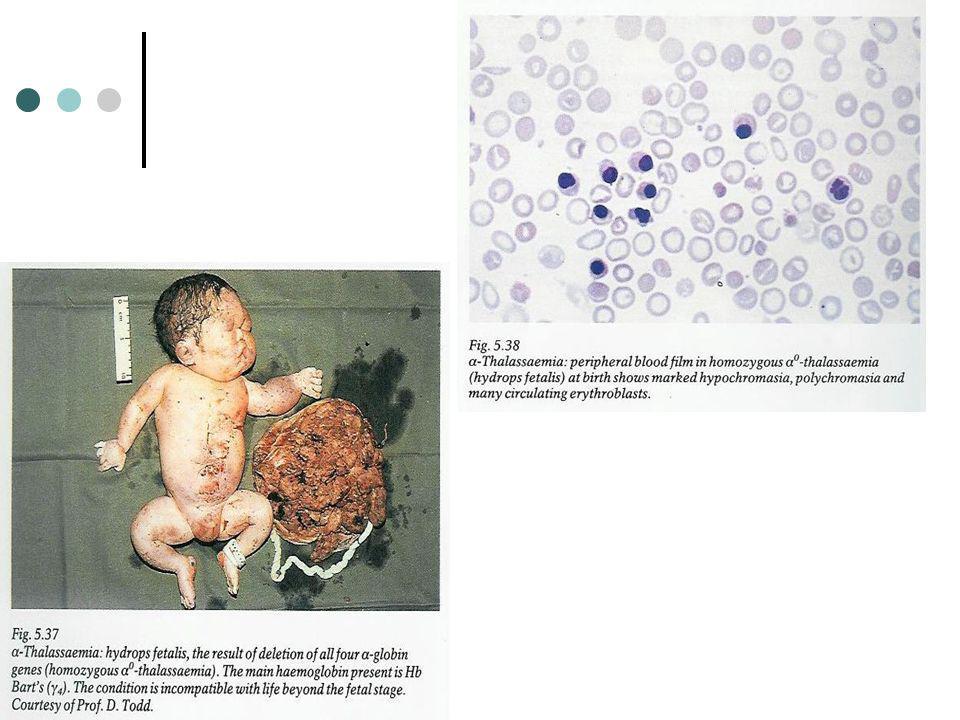
Quatro genes α sofreram delecção Não há produção de cadeias α Sudeste asiático Formação de tetrâmeros γ4 (Hb de Bart: 90-95%): alta afinidade para oxigénio » ø transporte de oxigénio aos tecidos) Hidrópsia com morte intrauterina ou ao nascimento (causada por anóxia tecidular, edema e falência cardíaca congestiva) Placenta edematosa e friável Hepatomegalia por tecido eritróide extra - medular Hb: g/dl Anisopoiquilocitose, hipocromia, GR nucleados Electroforese: Hb de Bart e Hb H Ø Hb A e Hb F Ambos os pais com talassemia α minor
: alta afinidade para oxigénio » ø transporte de oxigénio aos tecidos) Hidrópsia com morte intrauterina ou ao nascimento (causada por anóxia tecidular, edema e falência cardíaca congestiva) Placenta edematosa e friável. Hepatomegalia por tecido eritróide extra - medular. Hb: g/dl. Anisopoiquilocitose, hipocromia, GR nucleados. Electroforese: Hb de Bart e Hb H. Ø Hb A e Hb F. Ambos os pais com talassemia α minor.")
16
Prevenção: Filho de pais portadores ou com Hb H:
Análise de DNA fetal obtido por amniocentese ou biópsia das vilosidades coriónicas

17
Tratamento: Portadodes assintomáticos: Hb H: Aconselhamento genético
Transfusões Esplenectomia (se anemia excessiva)
")
18
Talassemia Beta

19
A talassemia beta resulta de mais de 150 diferentes mutações dos genes HBB responsáveis pela codificação das cadeias beta da globulina, resultando numa redução completa (βo talassemia) ou parcial (β+ talassemia) das mesmas. Portanto a extensão da delecção e da função dos genes não afectados determina a característica clínica dos diferentes genótipos. Ao contrário das talassemias alfa, a maior parte das talassemias beta são causados por mutações que afectam a regulação ou expressão génica ao invés de deçlecção génica. Os diferentes tipos de mutação têm expressões clínicas que variam de leves (minor) a graves (major).
 a graves (major).")
20
As mutações podem ser divididas naquelas que afectam a transcrição do mRNA ( mutação da região promotora e do codão de finalização) e naquelas que afectam o processamento do mRNA ( mutações de junções de splicing, novos sinais de splicing, clivagem e recombinação deficiente).
 e naquelas que afectam o processamento do mRNA ( mutações de junções de splicing, novos sinais de splicing, clivagem e recombinação deficiente).")
21
Talassemia major, Anemia de Cooley ou talassemia homozigótica deve-se á presença de duas mutações β talassémicas, idênticas uma em cada cromossoma 11. A heterogeneidade das β talassemias é tão vasta que a maior parte dos chamados homozigóticos são na verdade heterozigóticos para diferentes genes HBB que podem ocupar o mesmo loci. A talassemia intermédia caracteriza-se por ser uma doença moderada sem necessidades transfusionais de repetição. Geralmente é secundária a duas mutações β talassemicas A talassemia minor, também conhecida como traço talassémico ou talassemia β heterozigótica é devido á presença de uma única mutação pelo que o individuo produz normalmente duas cadeias alfa e uma beta.

22
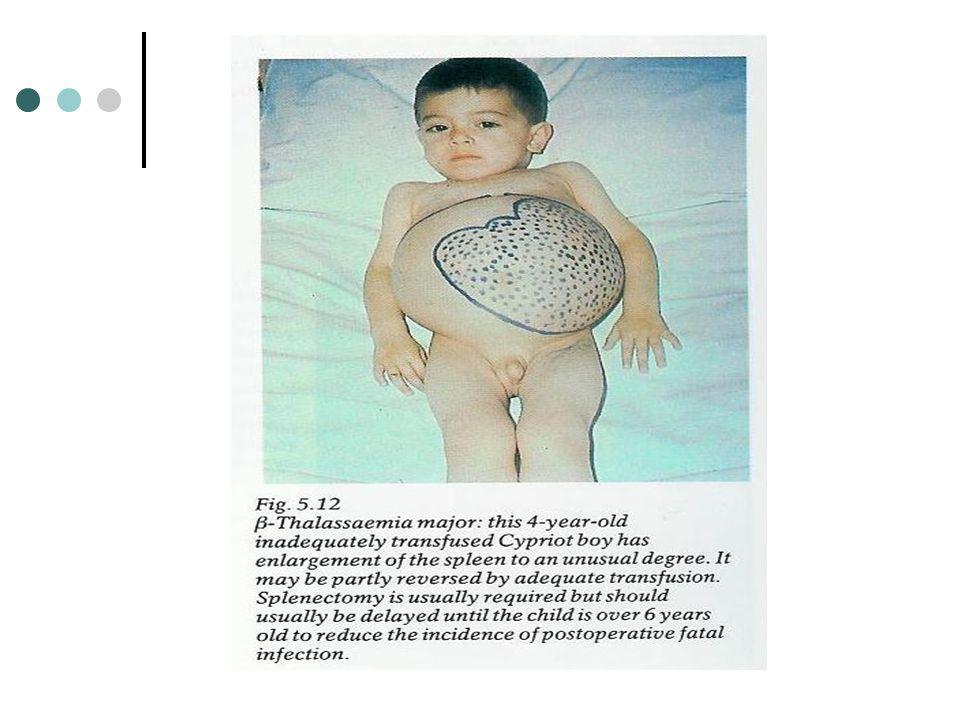
Talassemia beta major A βo talassemia encontra-se associada com a predominância de HB F, ausência de HbA e quantidades variáveis de Hb A2. A anemia e as alterações morfológicas dos eritrócitos são detectáveis às 6 semanas e a esplenomegalia às 8 semanas. Na ausência de terapêutica transfusional a concentração de Hb rapidamente cai para os 3 a 5 g/dl e as manifestações clínicas emergem. O diagnóstico pré-natal pode ser feito através da recolha de sangue fetal durante o segundo trimestre da gravidez

24
Alterações ósseas Aumento da reabsorção óssea
Diminuição da mineralização Expansão das cavidades medulares e adelgaçamento dos cortices metacarpo, metatarso e falanges com aspecto rectangular e convexo Expansão do diploe do cranio com tábuas interna e externa muito finas e com estriações entre ambas sugerindo na imagem radiográfica o aspecto de cabelo em pé

26
Atraso na pneumatização dos seios perinasais
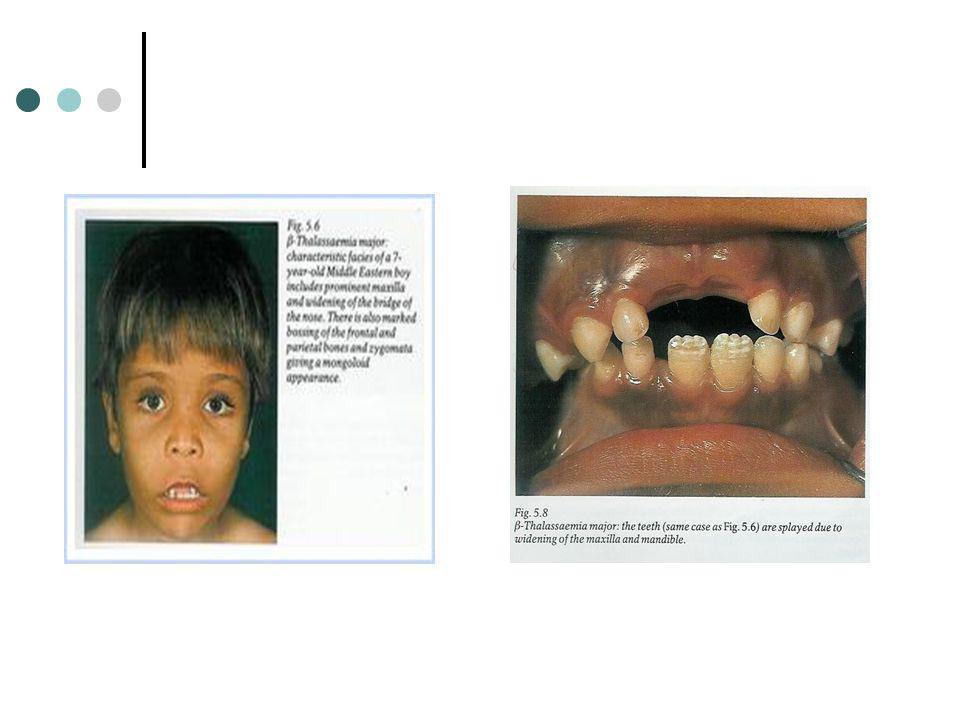
O crescimento exagerado da maxila produz maloclusões severas com tendência a obscurecer a base nasal e a expor os dentes superiores. Verifica-se ainda a presença de bossas frontais. Estes distúrbios do crescimento craniofacial dão origem ao Facies talassémico. Os ossos longos encontram-se predispostos para fracturas patológicas nomeadamente fracturas compressivas das vértebras Fusão prematura da epífise proximal do úmera ou epífise distal do fémur são frequentes.

28
Alterações do desenvolvimento e maturação sexual
O atraso no crescimento na infância precoce é consequência da anemia severa pelo que pode ser prevenida (mas não corrigida) por um programa agressivo de transfusões. Mesmo crianças optimamente transfundidas apresentam na adolescência crescimento atrasada ou encurtado. A puberdade encontra-se também atrasada e frequentemente incompleta. Os rapazes podem ter espermatogénese activa mas apresentam fraca libido. Diabetes mellitus e hipoparatiroidismo são situações frequentemente documentadas em doentes mais velhos, transfundidos, pelo que a sobrecarga de ferro se reveste da maior significância patogénica.
 por um programa agressivo de transfusões. Mesmo crianças optimamente transfundidas apresentam na adolescência crescimento atrasada ou encurtado. A puberdade encontra-se também atrasada e frequentemente incompleta. Os rapazes podem ter espermatogénese activa mas apresentam fraca libido. Diabetes mellitus e hipoparatiroidismo são situações frequentemente documentadas em doentes mais velhos, transfundidos, pelo que a sobrecarga de ferro se reveste da maior significância patogénica.")
30
Complicações cardiopulmunares
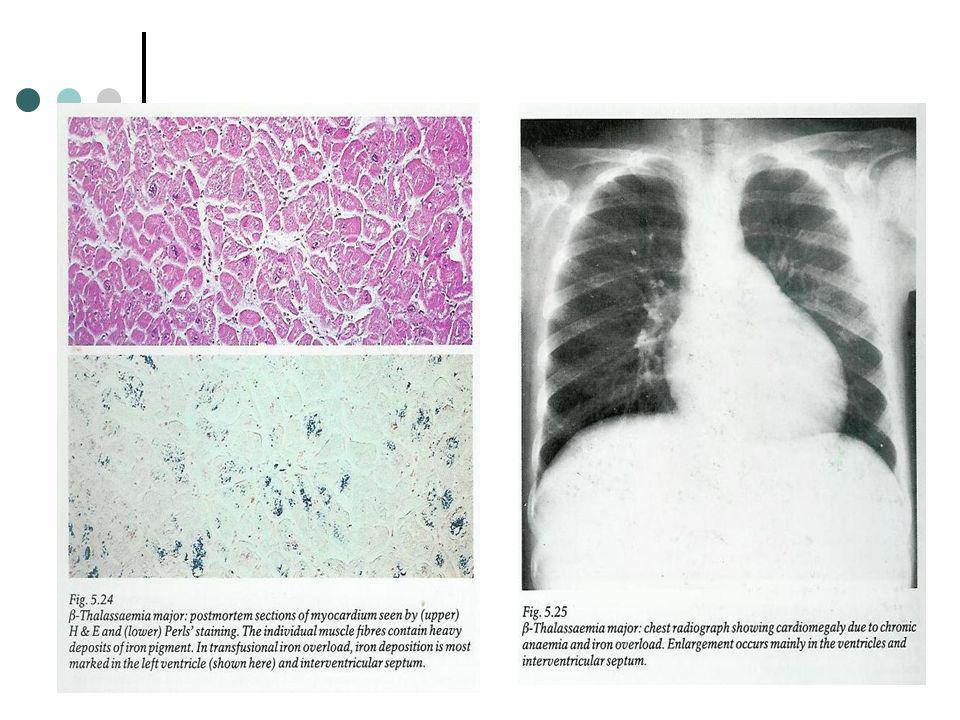
Após a primeira década muitos doentes experimentam pericardite estéril, como consequência de depósitos pericárdicos de ferro ou infecções por estirpes reumatológicas de streptococcus. A primeira cauda de morte nos doentes transfundidos é a Hemosiderose miocárdica que leva a arritmias e miocardipatia congestiva, normalmente a meio da segunda década de vida. A Introdução de iECA no tratamento de doentes com disfunção ventricular esquerda assintomática permitiu a prevenção e atraso da congestão cardíaca. A deposição pulmonar de ferro e o compromisso da expansibilidade diafragmática pelo crescimento esplénico concorrem para o obstrução aérea e hiperinsuflação dos pulmões.

32
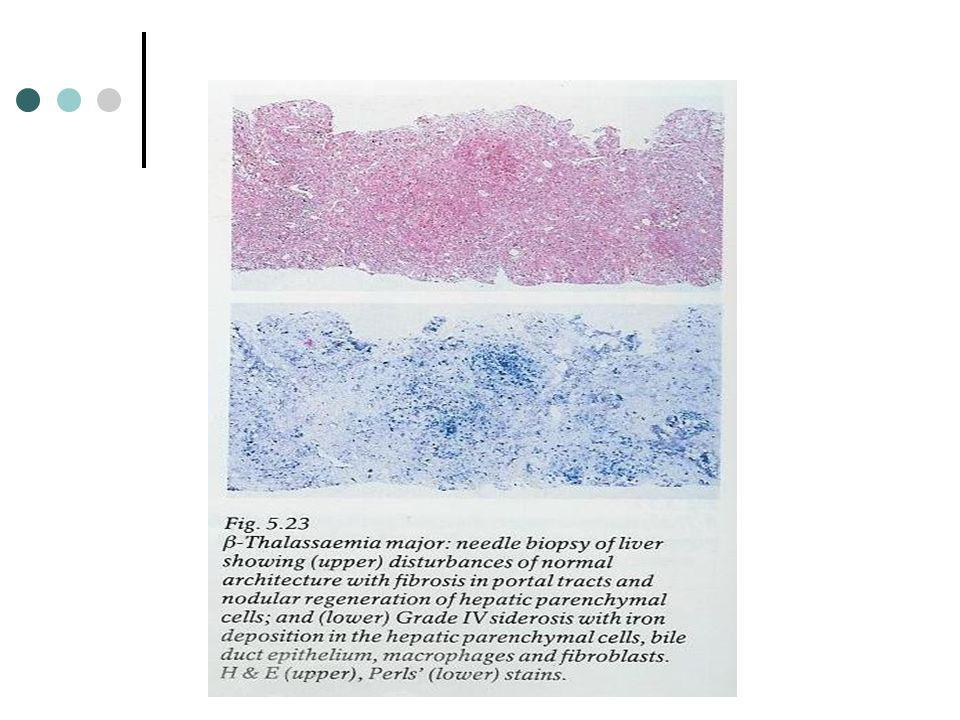
Doença hepatobiliar A expansão hepática no inicio da vida está relacionado com a hematopoiese extramedular. Mais tardiamente resulta de cirrose expansiva acompanhada de agregados nodulares de hepatócitos regenerados. O depósito de ferro envolve quer as células de Kupffer e hepatócitos produzindo um padrão indistinguível do da hemocromatose idiopática. Coaguloptia é causada pela falha na produção hepática de factores de coagulação embora hemorragia sintomática seja rara.

34
Apresentação hematológica
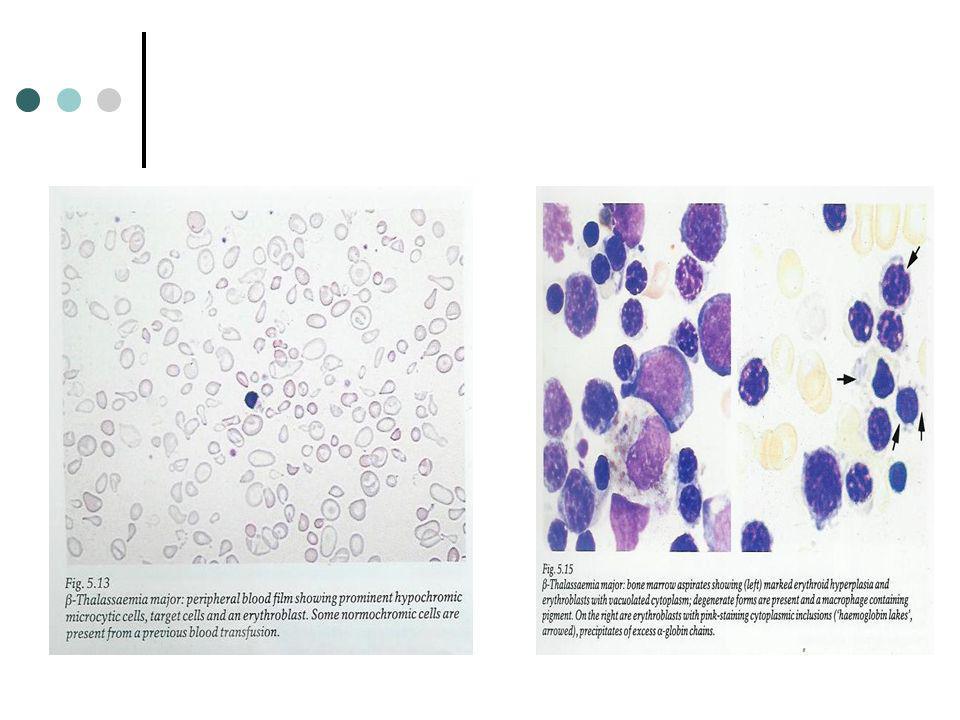
A anemia é geralmente pronunciada quando diagnosticada ( 2.5 a 6.5g/dl). A anemia é microcítica ( MCV 48 a 72fl) e hipocrómica (MCHC 23 a 32 g/dl). A anisocitose é marcada com a presença de células com diâmetros que vão desde os 3 aos 5 μm. Pontuado basófilo e presença de normoblastos pobremente pigmentados. Leucocitose resulta e um aumento de neutrófilos e em menor extensão da presença de mielócitos e metamielócitos. A medula óssea encontra-se incrivelmente hipercelular com profunda hiperplasia normoblastica. Aumento dos níveis de bilirrubina não conjugada e níveis séricos de ferro.
. A anemia é microcítica ( MCV 48 a 72fl) e hipocrómica (MCHC 23 a 32 g/dl). A anisocitose é marcada com a presença de células com diâmetros que vão desde os 3 aos 5 μm. Pontuado basófilo e presença de normoblastos pobremente pigmentados. Leucocitose resulta e um aumento de neutrófilos e em menor extensão da presença de mielócitos e metamielócitos. A medula óssea encontra-se incrivelmente hipercelular com profunda hiperplasia normoblastica. Aumento dos níveis de bilirrubina não conjugada e níveis séricos de ferro.")
36
Prognóstico O curso natural da talassemia caracteriza-se por infecções recorrentes, cacexia progressiva e morte por volta dos 5 anos. Com terapia transfusional, cerca de 25% dos doentes sobrevive até aos seus 20 anos. Ainda assim dois terços morrem com complicações cardíacas com a idade média de 16/17 anos. O ferro acumulado causa disfunção visceral quando excede 0.7g/kg e causa a morte quando atinge o 1g/kg.

37
Talassemia beta intermédia
Este síndrome é causado pela presença de um único alelo beta talassémico. É mais frequente na raça negra. O ratio de síntese γ:α é maior que nos pacientes com talassemia major e o pool de cadeias alfa livre é menor. As concentrações de Hb mantêm-se entre 6 a 9 g/dl sem ser submetido a transfusão. O sangue periférico apresenta alterações comparáveis com a talassemia major: anisocitose, hipocromia, células alvo, ponteado basófilo e numerosas formas nucleadas. A hiperplasia da medula óssea é proeminente e pode-se provar a presença de inclusões de cadeias alfa livres.

38
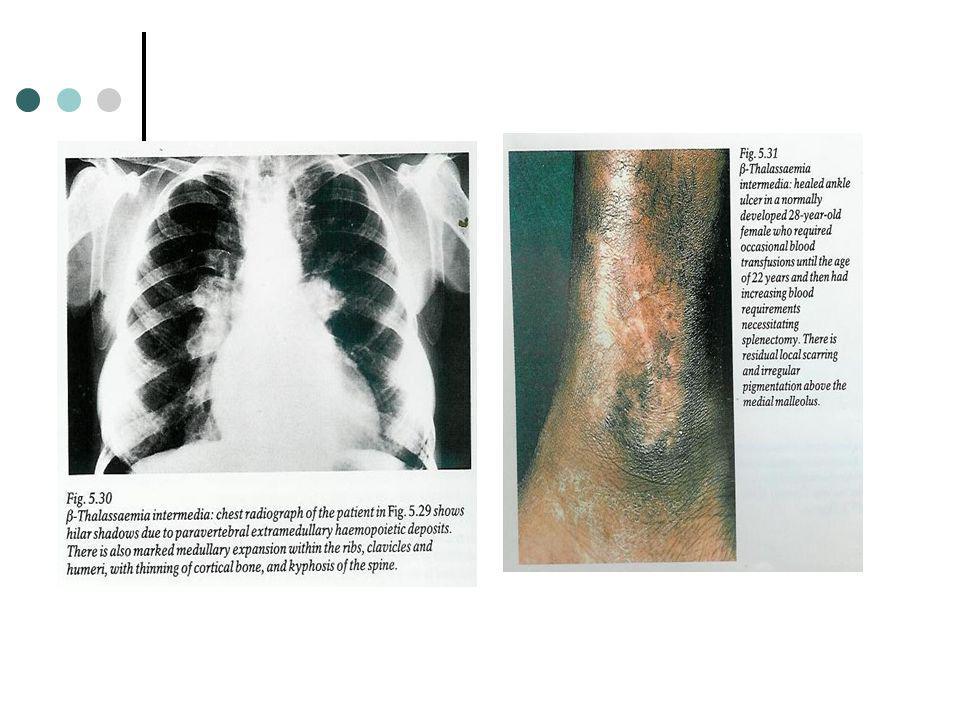
As manifestações clínicas da talassemia intermédia são comparáveis às da talassemia major mas de menor magnitude. Mesmo com anemia crónica, estes indivíduos não requerem transfusão a não ser quando existe uma patologia infecciosa concomitante. O crescimento e desenvolvimento durante a infância encontra-se relativamente descomprometido, a puberdade não é afectada e a fertilidade é preservada. Regularmente observa-se palidez, icterícia intermitente, esplenomegalia e alterações craniofaciais. Complicações na vida adulta incluem fracturas patológicas, coleolitíase e massas torácicas correspondentes a tecido hematopoiético.

40
Talassemia minor Este síndrome é descoberto a maior parte das vezes durante exames de rotina em doentes assintomáticos apresentando valores aumentado de HbA2 ou HbF. Clinicamente manifesta-se por icterícia, esplenomegalias, úlceras de perna ou achados radiológicos nos ossos longos. As mulheres afectadas tornam-se mais anémicas durante a gravidez não sendo no entanto necessário recorrer a transfusões. A anemia é mediana ou ausente mas as alterações morfológicas do sangue periférico são proeminentes: microcitose, hipocromia, anisocitose, células alvo e pontuado basófilo. De destacar a ausência de células vermelhas nucleadas. A contagem de reticulócitos e de bilirrubina sérica pode estar ligeiramente elevada. A medula óssea apresenta discreta hiperplasia eritroide sem presença de inclusões de cadeias alfa nas células percursoras.

41
Talassemia minima Trata-se duma substituição pontual no codão 24.
Refere-se à condição em que a alteração na síntese de cadeias beta é tão subtil que escapa a uma avaliação clínica ou hematológica convencional. Daí poder também denominar-se de talassemia silenciosa. Trata-se duma substituição pontual no codão 24. Os valores de MCV e MCH são normais ou ligeiramente diminuídos e o padrão da hemoglobina não apresenta alterações O diagnóstico só pode ser feito recorrendo ao estudo da síntese das cadeias da globulina, que revelam um ratio beta:alfa de 1.3.

42
Terapêuticas Transfusão
Os programas de hipertransfusão suprimem efectivamente a actividade eritroide prevenindo a expansão ilimitada da medula óssea responsável pelas alterações ósseas características da talassemia major. As transfusões regulares devem começar quando o valor da concentração da Hb é inferior a 7g/dl ou o crescimento se encontre comprometido. O crescimento fica permanentemente comprometido se os níveis de Hb não forem mantidos estáveis até aos 3 a 4 anos de idade. São vantagens adicionais deste programa a prevenção ou atraso do desenvolvimento de esplenomegalia congestiva, infecção recorrente e diminuição da reserva cardíaca.

43
Contudo, a terapêutica transfusional pode levar a infecções transmitidas por via sanguínea, aloimunização, reacções febris e letal overdose de ferro. Uma unidade de sangue transfundido equivale ao intake anual de ferro. Os doentes que recebem mais de 100 unidades de sangue transfundido apresentam grande probabilidade de desenvolver hemosiderose. Tornou-se então imperativo a quelação do ferro recorrendo a agentes quelantes tais como a desferoxamina (Desferal®) que para apresentar resultados significativos na sobrevivência do doente tem que começar a ser administrado antes dos 5 anos de idade no caso da talassemia beta major.
 que para apresentar resultados significativos na sobrevivência do doente tem que começar a ser administrado antes dos 5 anos de idade no caso da talassemia beta major.")
44
Esplenectomia Como explicado anteriormente, um correcto programa de transfusões retarda o desenvolvimento de esplenomegalia sem no entanto evitar o sequestro de células transfundidas. O casionalmente o sequestro esplénico de plaquetas e leucócitos é suficiente para produzir trombocitopenia e leucopnia. A maior indicação para a realização de esplenectomia é o aumento das necessidades transfusionais.

45
Transplante de medula Terapia génica
Esta terapêutica providencia células totipotentes capazes de expressar Hb normal. Se for aplicada antes dos órgãos alvos serem seriamente afectados, o transplante é curativo em cerca de 80 a 90% dos casos. Terapia génica O uso de vectores capazes de introduzir o gene correcto nas células hematopoiéticas percursoras revelou-se incapaz. A promoção da síntese de Hb F tem como objectivo melhorar os sintomas da beta talassemia. Para tal recorre-se a administrações intermitentes de butirato sem no entanto se encontrar resultados conclusivos e esclarecedores da aplicação desta esperança terapêutica.

46
Talassemia δβ

47
Diminuição ou ausência da síntese de cadeias das globulinas β e δ
Aumento compensatório da produção de cadeias γ Aumento de HbF ( praticamente 100%da hemoglobina em homozigóticos) Maioria resulta de delecções envolvendo toda ou porções das cadeias δ e β mas poupando os 2 tipos de cadeias γ (GγAγ) Existem delecções que envolvem também a cadeia Aγ Fenótipo tipo β0 ( uma pequena minoria em que o defeito na síntese de cadeias β é por mutação e não delecção pode ter fenótipo β+)
 Maioria resulta de delecções envolvendo toda ou porções das cadeias δ e β mas poupando os 2 tipos de cadeias γ (GγAγ) Existem delecções que envolvem também a cadeia Aγ. Fenótipo tipo β0 ( uma pequena minoria em que o defeito na síntese de cadeias β é por mutação e não delecção pode ter fenótipo β+)")
48
Talassemia γδβ

49
Forma rara de talassemia
Delecção ou inactivação de todo o complexo do gene β por distúrbios na conformação da cromatina por delecção ou por perda de sequências 5’ necessárias para a expressão do gene das cadeias β in vivo Sem forma homozigótica encontrada até hoje Caracterizada por anemia hemolítica severa neonatal que progressivamente evolui para talassemia β minor

50
Caso clínico Talassemia β major

51
identificação Nome :M.J.S.A. Idade :3 anos
Data de nascimento : Sexo : ♂ Raça : Caucasiana Etnia: Cigana Naturalidade :Guimarães Residência : Guimarães

52
Motivo de internamento
Quadro de anemia grave não especificada

53
Hx da doença actual Admitido no S.U. do H.S.O. em Março de com um quadro de febre Exame objectivo: palidez cutaneo-mucosa marcada febre 39ºC Exames complementares de dx: rastreio séptico ( resultados não conhecidos) hemograma: anemia grave: Hg 5.6g/dl pancitopenia ( transferência para o H.S.J.)
 hemograma: anemia grave: Hg 5.6g/dl pancitopenia. ( transferência para o H.S.J.)")
54
Hx da doença actual Já no HSJ (21-03-03)
Exame objectivo: Palidez cutâneo mucosa Icterícia Esplenomegalia Hepatomegalia Taquicardia

55
Doente fica internado para estudo
Hx da doença actual Repetição do hemograma: Anemia grave: 5.5g/dl Sem atingimento das restantes séries hematológicas Observação do esfregaço sanguíneo: compatível com o dx de talassemia Doente fica internado para estudo

56
Hx da doença actual Terapêutica: Vigilância Transfusão de GR

57
Hx da doença actual Durante todo o internamento o doente esteve hemodinamicamente estável e apirético. Boa tolerância alimentar

58
Hx da doença actual Após a 1ª transfusão( na admissão)
Hemograma:Hg-7.1g/l esfregaço: 27eritroblastos/100cel nucleadas Ht-22% policromatófilia VGM- 69fl anisopoilocitose + + + células em alvo + pontuado basófilo + microcitose + + hipocromia + +

59
Hx da doença actual Prova de Combs directa positiva
???????????????????????????

60
Hx da doença actual Exames complementares de dx requisitados: V.D.R.L.
T.P.H.A. Toxoplasmose Rubéola VHS 1 e2 CMV Estudo analítico dos pais Ecografia abdominal

61
Hx da doença actual Hemograma(24-03-03): Hg-8.1g/l Ht-24.3%
VGM- 69.9fl

62
26-03-03: nova transfusão de GR
Hx da doença actual : Serologia de sangue para doença por citomegalovirus: Atc IgG – positivo (144.8 AU/ml) Atc IgM – duvidoso ( pedido de estudo molecular) Serologias IgM IgG Toxoplasmose - Rubéola VHS1 VHS2 CMV duvidoso + ecografia abdominal: aumento do tamanho do fígado e baço. Sem alterações da arvore biliar Reacção do V.D.R.L. – negativa Reacção de T.P.H.A – negativa : nova transfusão de GR
 Atc IgM – duvidoso. ( pedido de estudo molecular) Serologias. IgM. IgG. Toxoplasmose. - Rubéola. VHS1. VHS2. CMV. duvidoso. + ecografia abdominal: aumento do tamanho do fígado e baço. Sem alterações da arvore biliar. Reacção do V.D.R.L. – negativa. Reacção de T.P.H.A – negativa : nova transfusão de GR.")
63
Hx da doença actual Hemograma e esfregaço dos progenitores: ♂ ♀
Hg (g/l) 12 11.2 Ht (%) 38.4 36 VGM (fl) 68 61 anisocitose + microcitose ++ hipocromia hemograma esfregaço Alterações compatíveis com o dx de talassemia β minor
 Ht (%) VGM (fl) anisocitose. + microcitose. ++ hipocromia. hemograma. esfregaço. Alterações compatíveis com o dx de talassemia β minor.")
64
Dx de talassemia β major
Hx da doença actual Pedido do estudo molecular do lactente e pais: Mãe: heterozigótica para a mutação IVS1-110 no gene que codifica para a cadeia β da Hg Pai: heterozigótico para a mutação IVS1-110 no gene que codifica para a cadeia β da Hg Lactente: homozigótico para a mutação IVS1-110 nos genes que codifica para a cadeia β da Hg Dx de talassemia β major

65
Hx da doença actual Hemograma dia 28.03.03 Hg-9.1g/l Ht-29.7%
VGM- 62.4fl Biologia molecular de sangue: DNA Vírus Citomegálico positivo

66
Hx da doença actual Doente tem alta dia 1 de Abril de 2003 sendo tratado em ambulatório e seguido nas consultas de hematologia pediátrica. Faz desde então 2 a 3 transfusões de GR por semana Começou desde os dois anos de idade a fazer desferoxamina (quelante do ferro) Durante estes 3 anos não teve nenhuma patologia de relevo.
 Durante estes 3 anos não teve nenhuma patologia de relevo.")
67
Hx da doença actual/ perspectivas futuras
Doente candidato a transplante de medula durante o ano corrente

68
Antecedentes pessoais
Gestação de termo, não vigiada Parto eutócico, hospitalar (HSO), choro imediato ao nascimento Antropometria 3042/48 Apgar: 9/10 (sem necessidade de reanimação) Aleitamento materno até 7 meses PNV actualizado Sem alergias ou reacções adversas a fármacos conhecidas Sem antecedentes patológicos de relevo (sarampo, rubéola, parotidite) Sem cirurgias anteriores Transfusões sanguíneas semanais (2 a 3 por semanas)
, choro imediato ao nascimento. Antropometria 3042/48. Apgar: 9/10 (sem necessidade de reanimação) Aleitamento materno até 7 meses. PNV actualizado. Sem alergias ou reacções adversas a fármacos conhecidas. Sem antecedentes patológicos de relevo (sarampo, rubéola, parotidite) Sem cirurgias anteriores. Transfusões sanguíneas semanais (2 a 3 por semanas)")
69
Hx social Pai e mãe sem escolaridade Pai pedinte
Residiam até há pouco tempo numa barraca, sem água, luz ou saneamento básico com muito más condições de higiene Actualmente vivem num apartamento cedido pela câmara municipal com luz, água e saneamento básico mas mantendo se as más condições de higiene Condições económicas precárias

70
Restantes antecedentes familiares irrelevantes
Hx familiar ♂ ♀ A a A AA Aa I a Aa aa II III I,1 - bócio II, anos, talassemia β minor II, anos, talassemia β minor III,1 - 7 anos, talassemia β minor III,2 - 5 anos, talassemia β minor III, meses saudável Restantes antecedentes familiares irrelevantes

71
Revisão por aparelhos e sistemas
Geral: refere astenia fatigabilidade desde que começou a andar. Refere febre não quantificada desde há dois dias. Nega alterações do apetite e do peso habitual. Pele: palidez cutânea “desde sempre”. Nega alterações da coloração da pele assim como eritema, prurido. Nega alterações do cabelo e unhas. Olhos: Nega uso de óculos e outras alterações da visão. Nega fotofobia, diplopia, infecções oculares, glaucoma ou traumatismos Ouvidos: Acuidade auditiva sem alterações. Nega vertigens e tinitos. Sem hx de infecções e otorreia Nariz: rinorreia amarelo esverdeado desde há dois dias. Nega alterações do olfacto e epistáxis. Sem hx de sinusite ou traumatismo Garganta e boca: Nega exsudados na orofaringe ou faringites. Nega alterações do paladar e voz.

72
Revisão por aparelhos e sistemas
Endócrino: nega polidipsia, poliúria ou polifagia. Nega alterações de tamanho ou dor na região tiroidea. Nega alterações do peso, apetite, sudação e intolerância ao frio/calor. Pulmonar: refere uma tosse profunda produtiva desde há dois dias. Expectoração amarelada, factos para os quais não foi medicado até à data. Nega hemoptises, dispneia, dor torácica ou pieira. Sem história de tuberculose, asma ou bronquite Cardiovascular: nega palpitações, dor torácica ou claudicação intermitente. Nega dispneia de esforço ou ortopneia. Nega edemas dos membros, tromboflebites ou úlceras Hematológico: nega tendência para hematomas e sangramento. Quadro de anemia grave (referido da hx da doença actual). Transfusões sanguíneas semanais desde os 4 meses até à data.
. Transfusões sanguíneas semanais desde os 4 meses até à data.")
73
Revisão por aparelhos e sistemas
Gastrointestinal: nega náuseas e alterações do trânsito intestinal. Nega disfagia, vómitos, hematemeses, rectorragias, diarreia ou alterações na coloração das fezes. Sem hx de hepatite e doença biliar Génito-urinario: nega alterações da coloração da urina. Nega disúria, noctúria, hematúria, poliúria assim como dor no flanco ou supra púbica. Músculo esquelético: nega dores e alterações/ deformidades ósseas. Sem alterações da força ou mobilidade muscular. Sem dor ou rigidez nas articulações

74
Revisão por aparelhos e sistemas
Linfático: nega adenomegalias Neurológico: nega quedas, tonturas, síncope, convulsões, paralisias, alterações da coordenação ou sensitivas. Nega hx de traumatismo craneo-encefálico. Psiquiátrico: refere irritabilidade e insónias desde o nascimento. Nega alterações de memória ou comportamentais.

75
Exame objectivo Doente em bom estado geral, consciente, muito pouco colaborante, orientado no espaço e no tempo. Bom estado nutricional Idade aparente coincidente com a real Sem sinais de sofrimento agudo -

76
Exame objectivo Antropometria: Sinais vitais: Peso:12kg Altura:82cm
IMC:17.9 Sinais vitais: Temperatura: 38ºC F.C.: 80bt/s F.R.: 21 ciclos por segundo T.A.: não foi medida por ma colaboração do doente

77
Exame objectivo Pele e faneras: palidez e xerose cutânea. Temperatura e textura normais. Sem edema. Ausência de lesões primárias e secundarias na pele. Distribuição pilosa normal, unhas de configuração normal e sem alterações do leito ungueal. Cabeça: Inspecção: craneo de configuração normal, sem dismorfias ou tumefacções. Implantação do cabelo normal e configuração da mimíca facial Palpação: sem lesões ou pontos dolorosos no couro cabeludo. Artérias temporais palpáveis simétricas e sem dor à palpação

78
Exame objectivo Olhos: Pálpebras simétricas e móbeis, sem sinais inflamatórios. Arcada super ciliar com configuração e distribuição pilosa normal. Conjuntiva de coloração normal e sem sinais inflamatórios. Esclera ligeiramente ictérica. Ouvidos: pavilhões auriculares com configuração e implantação normais. Sem otorreia. Sem adenomegalias retroauriculares. Fossas nasais: pirâmide e septo nasal de configuração normal. Rinorreia abundante amarelo/esverdeado. Sem epistáxis. Sem dor sobre os seios peri nasais. Cavidade oral e oro-faringe: lábios de coloração normal, sem lesões. Língua de tamanho e configuração normais sem ulcerações. Mucosa oral e gengivas de coloração normal e sem lesões. Amígdalas de tamanho e configuração normal sem exsudados

79
Exame objectivo Pescoço: configuração normal e sem tumefacções visíveis. Sem adenomegalias cervicais ou supra claviculares. Glândula parótida e submaxilar de dimensões normais sem nódulos ou dor à palpação. Tiróide de forma e tamanho normal, sem nódulos ou pontos dolorosos. Tórax: simétrico com diâmetro transverso maior que o antero posterior. Sem tumefacções ou massas palpáveis Respiratório: Inspecção: expansibilidade normal da caixa torácica. Movimentos respiratórios simétricos. Sem uso de movimentos acessórios Palpação: movimentos simétricos. Transmissão normal e simétrica das vibrações vocais. Percussão: ressonância em toda a área pulmonar sem pontos de Macicez Auscultação: sons respiratórios preservados e simétricos. Sem ruídos adventícios. Relação inspiração/expiração normal

80
Exame objectivo Sistema cardiovascular: pressão arterial não determinada. Pulso arterial radial amplo, rítmico, regular e simétricos. Restantes pulsos arteriais palpáveis e com as mesmas características. sem turgescência venosa jugular nem refluxo hepato-jugular. Sem sinais de edema. Membros inferiores com temperatura, cor e distribuição pilosa normal sem distensão venosa. F.C.- 80 btm/s Coração: Inspecção: Ausência de pulsatibilidades, área de impulso máximo não visível Palpação: área de impulso máximo palpável com localização normal. Ausência de frémitos ou lifts Auscultação: 1º e 2º som audível em todos os focos e sem alterações. Ausência de sopros ou sons adicionais.

81
Exame objectivo Abdómen:
Inspecção: configuração normal, simétrico, sem circulação venosa visível, cicatriz umbilical normal e pigmentação normal Palpação: superficial e profunda sem dor. Sem tumefacções palpáveis. Fígado e baço palpáveis com tamanho normal Macicez normal na área hepática com tamanho do fígado normal. Timpanismo normal na área gástrica. Auscultação: sons intestinais audíveis com características normais. Ausência de sopros ( Não foi realizado o exame objectivo do aparelho genital e neurológico)
")
82
Hemograma 21-03-03 24-03-03 28-03-03 Eritrócitos (x1012 /l) 3.24 3.58
4.10 Hemogbina (g/l) 7.2 8.8 9.8 Volume globular % 21.3 24.3 29.7 MCV (fL) 65.7 67.9 72.4 MCH (pg) 22.2 22.6 23.9 MCHC (g/dl) 33.8 33.3 33.0 RCW-CV (%) 29.9 30.9 26.2 RDW-SD (fL) 73.6 77.9 72.5 Leucócitos (x109/l) 8.55 10.22 8.31 Neutrófilos (%) 27.5 27.7 Eosinófilos (%) 1.4 2.6 2.8 Basófilos (%) 2.5 0.8 0.2 Linfócitos (%) 56.6 60.1 51.5 Monócitos 12.0 12.5 Plaquetas (x109/l) 204 382 Electoforese Hgs Barra acentuada HbF HbA2 (%) 2.7% HbF(%) 60.6% Anisopoiquilocitose moderada, policromatofilia Anisopoiquilocitose moderada, policromatofilia
 Volume globular % MCV (fL) MCH (pg) MCHC (g/dl) RCW-CV (%) RDW-SD (fL) Leucócitos (x109/l) Neutrófilos (%) Eosinófilos (%) Basófilos (%) Linfócitos (%) Monócitos Plaquetas (x109/l) Electoforese Hgs. Barra acentuada HbF. HbA2 (%) 2.7% HbF(%) 60.6% Anisopoiquilocitose moderada, policromatofilia. Anisopoiquilocitose moderada, policromatofilia.")
83
Quimica geral AST U/L 35 33 36 34 40 31 ALT U/L 12 14 15 16 13 18 21 Desidrogenase lactica U/L 588 293 372 283 284 304 Glicose g/L 0.82 0.80 0.76 0.75 0.87 0.81 Ureia g/L 0.11 0.14 0.08 0.09 0.23 0.26 0.25 Creatinina mg/L 2.6 3.4 3.0 2.8 4.1 Bilirrubina total mg/L 11.5 16.7 12.3 9.4 10.9 18.6 13.1 Bilirrubina directa mg/L 2.7 3.8 2.2 4.4 Ferro g/dl 154 Transferritina mg/dl 313 Ferritina ng/ml 83.62 225.15 487.38 2440 1770 2141

84
Resumo M.J.S.A., sexo ♂, 3 anos, deu entrada no S.U.em Março de 2003 com um quadro de anemia grave (Hg 5.5 g/dl), hepato e esplenomegalia e observação do esfregaço sugestiva de talassemia. Faz transfusão de GR na admissão com resposta positiva do valor de Hg que sobe para 7.2 g/dl Fica internado por 12 dias, durante os quais faz mais 1 transfusão Estudo analítico e molecular dos pais e do lactente revela o dx de talassemia β major para o menor Realização de transfusões sanguíneas desde a alta até à alta. Terapêutica com deferoxamina desde os 2 anos A terapêutica revela boa resposta com valores de Hg sempre dentro da normalidade durante estes anos Candidato a transplante de medula durante o presente ano.
, hepato e esplenomegalia e observação do esfregaço sugestiva de talassemia. Faz transfusão de GR na admissão com resposta positiva do valor de Hg que sobe para 7.2 g/dl. Fica internado por 12 dias, durante os quais faz mais 1 transfusão. Estudo analítico e molecular dos pais e do lactente revela o dx de talassemia β major para o menor. Realização de transfusões sanguíneas desde a alta até à alta. Terapêutica com deferoxamina desde os 2 anos. A terapêutica revela boa resposta com valores de Hg sempre dentro da normalidade durante estes anos. Candidato a transplante de medula durante o presente ano.")
Apresentações semelhantes













, e que pode levar a incapacitação funcional.>")



 Reação mais comum durante a transfusão Motivo:>")



