Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Fibrose Cística Professora: Elisângela de Paula
Universidade Federal de Goiás Faculdade de Medicina Genética Fibrose Cística Professora: Elisângela de Paula Alunos participantes: Rafael Inagaki Thaís Teles Thaís de Toledo

2
Conceito A fibrose cística é uma doença genética letal, de herança autossômica recessiva. Ela faz certas glândulas produzirem secreções anormais, resultando disso vários sintomas, os mais importantes afetando o trato digestivo e os pulmões

3
Gen afetado O gen DF508 controla a produção de uma proteína que regula a transferência de cloreto e sódio através das membranas celulares. Quando há homozigose, a transferência de sódio e cloreto é defeituosa, conduzindo a desidratação e ao aumento de viscosidade das secreções.

4
Para melhor orientação há livros de fácil acesso a respeito da fibrose cística.

5
Diagnóstico O teste de suor, confirma o diagnóstico. O resultado é positivo quando temos mais de 60 ml/l de cloro na amostra colhida de suor da criança e 80 ml /l no adulto.

6
Apesar de muitos portadores desconhecerem que possuem a doença, associações estão sendo formadas com a finalidade de orientar e contribuir para uma melhoria na qualidade de vida desses pacientes.

7
Aproximadamente metade das crianças com fibrose cística são levadas ao médico pela primeira vez por apresentarem tosse persistente, sibilos, ou infecções respiratórias de repetição. A tosse, o sintoma mais evidente, é freqüentemente acompanhada por engasgos, vômitos e distúrbios do sono.

8
Mudanças nas glândulas
Nas glândulas dos intestinos e do pâncreas as secreções são grossas ou sólidas e podem bloquear completamente as glândulas. As glândulas produtoras de muco dos pulmões produzem secreções anormais que entopem as vias aérea.

9
Secreções brônquicas espessas começam obstruindo as pequenas vias aéreas que inflamam.
Com a progressão da doença, as paredes brônquicas engrossam, as vias aéreas se enchem de secreções infectadas, áreas do pulmão colabam (uma condição chamada atelectasia), e os linfonodos aumentam.
, e os linfonodos aumentam.")
10
Com a progressão da doença:
o tórax adquire a forma de barril, oxigênio insuficiente pode fazer os dedos ficarem alargados (como baquetas de tambor) pele ficar azulada. pólipos podem surgir no nariz. os seios para-nasais podem se encher de secreções espessas.
 pele ficar azulada. pólipos podem surgir no nariz. os seios para-nasais podem se encher de secreções espessas.")
11
os adolescentes em geral têm atraso na puberdade.
as complicações incluem pneumotórax, tosse com sangue, insuficiência cardíaca e função reprodutiva prejudicada. bronquites recorrentes e pneumonias destroem gradualmente os pulmões. alguns desenvolvem diabete insulino-dependente porque o pâncreas lesado não produz insulina suficiente.

12
o bloqueio de dutos biliares por secreções espessas pode conduzir à inflamação do fígado e, eventualmente, à cirrose hepática. a cirrose pode causar hipertensão porta, provocando varizes esofágicas (hemorragia). a morte geralmente resulta de uma combinação de insuficiência respiratória e insuficiência cardíaca, causadas pela doença pulmonar subjacente.
. a morte geralmente resulta de uma combinação de insuficiência respiratória e insuficiência cardíaca, causadas pela doença pulmonar subjacente.")
13
Campanha de conscientização sobre a fibrose cística

14
Pesquisa de Casos Em um período de 20 anos, 127 pacientes portadores de FC foram acompanhados longitudinalmente e submetidos a protocolo previamente estabelecido, após confirmação do diagnóstico pelo teste do suor. O genótipo foi obtido de 106 pacientes pela técnica do PCR. Os pacientes foram seguidos por mediana de 44 meses.

15
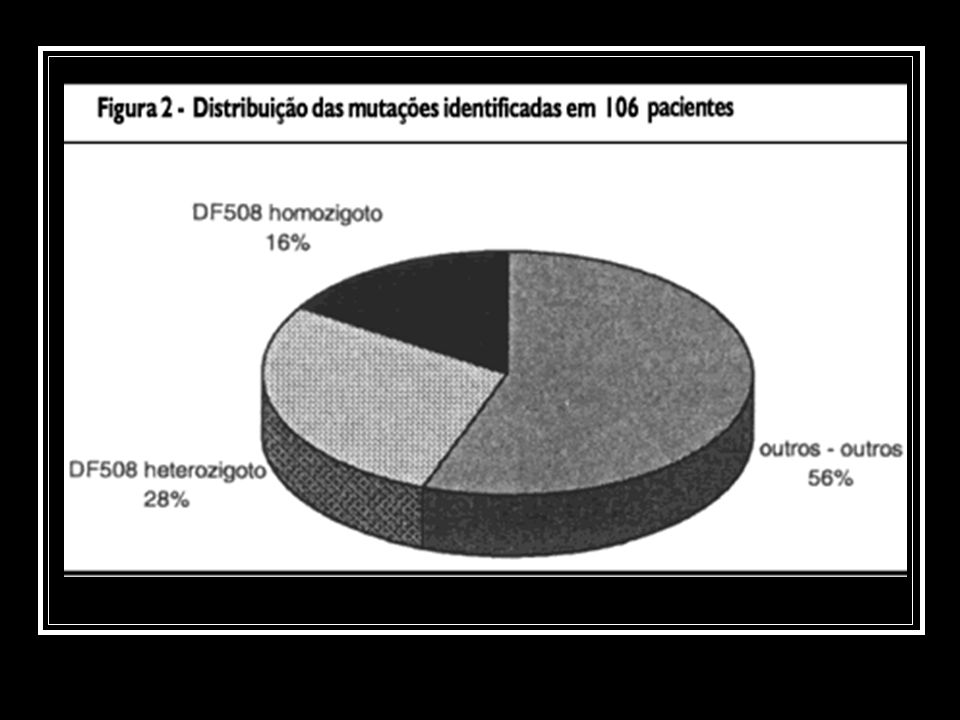
212 cromossomos estudados,
a mutação mais freqüente foi a DF508, encontrada em 64 cromossomos (30%), Os pacientes foram agrupados em três categorias com relação à presença ou não da mutação mais comum na FC: homozigoto, heterozigoto e outras mutações. Dezessete pacientes (16%) foram homozigotos para a mutação DF508; 30 pacientes (28%), heterozigotos; 59 (56%) não apresentaram a mutação DF508.
, Os pacientes foram agrupados em três categorias com relação à presença ou não da mutação mais comum na FC: homozigoto, heterozigoto e outras mutações. Dezessete pacientes (16%) foram homozigotos para a mutação DF508; 30 pacientes (28%), heterozigotos; 59 (56%) não apresentaram a mutação DF508.")
17
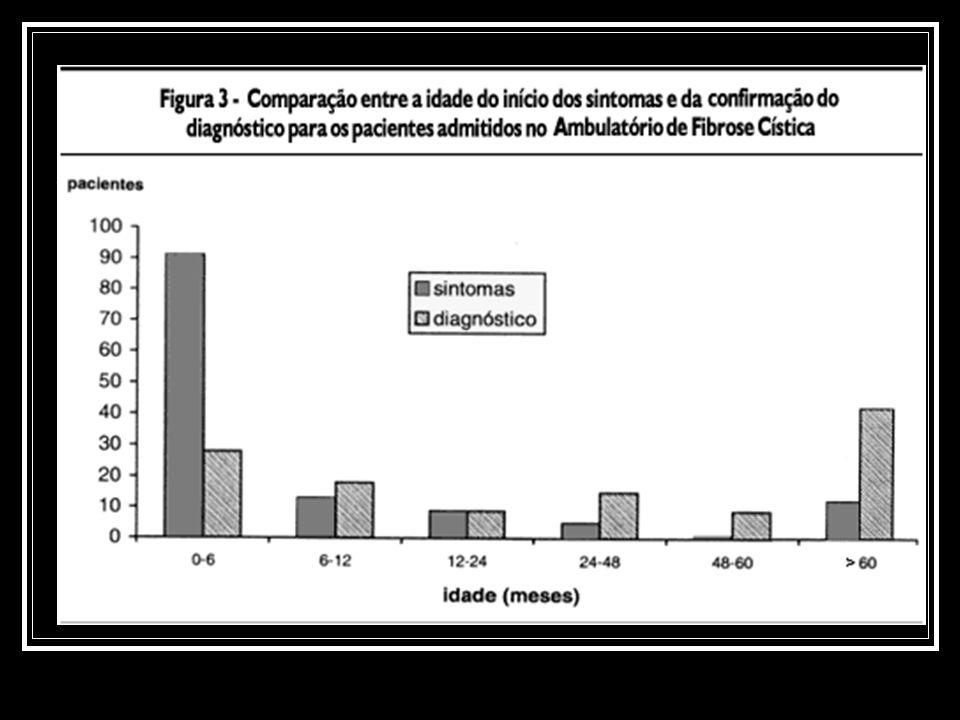
A idade de início dos sintomas variou de 1 a 186 meses
A idade de início dos sintomas variou de 1 a 186 meses. Um total de 91 pacientes (72%) já apresentavam sintomas nos primeiros 6 meses de vida. A média de idade ao diagnóstico foi de 57 ± 62,2 meses.
 já apresentavam sintomas nos primeiros 6 meses de vida. A média de idade ao diagnóstico foi de 57 ± 62,2 meses.")
19
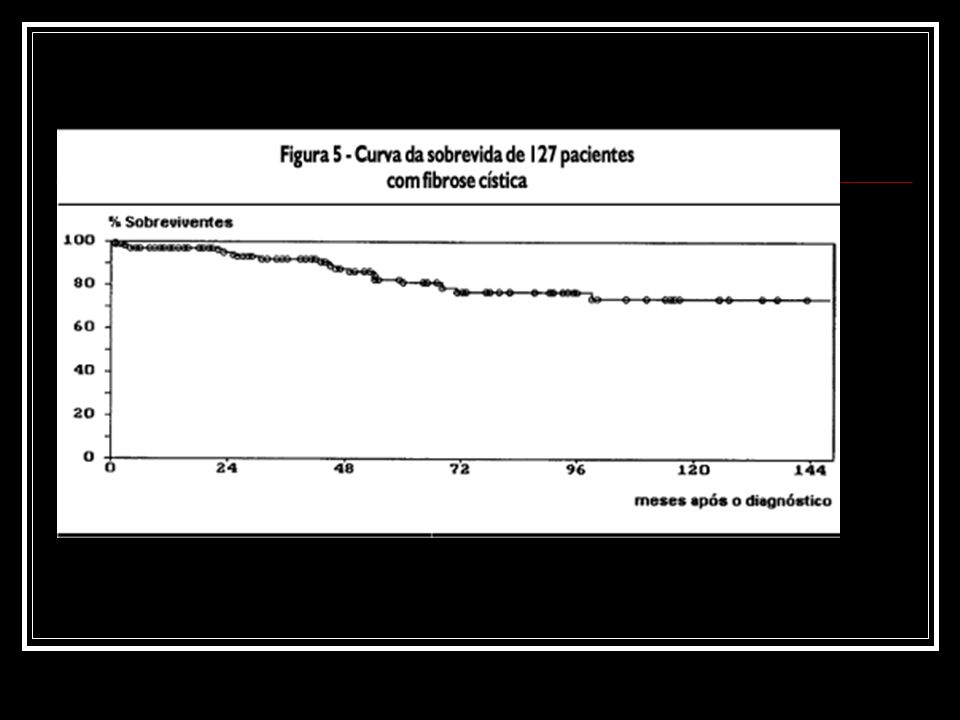
No tempo de seguimento abrangido pelo estudo, 22 (17,3%) dos 127 pacientes evoluíram para a insuficiência respiratória. Dos 127 pacientes incluídos na amostra, 20 (15,7%) evoluíram para o óbito no tempo de seguimento.
 evoluíram para o óbito no tempo de seguimento.")
Apresentações semelhantes



 REDUZIR RISCO DE TRANSMISSÃO>")







>")








 ou mucoviscidose é uma doença autossômica recessiva caracterizada por secreções anormalmente densas das glândulas mucosas,>")