Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Doença de Parkinson Vítor Vieira Piseta Curitiba, 2014

2
HISTÓRICO 1817 – James Parkinson publica o ensaio “An Essay on the Shaking Palsy”. Final do século XIX – Descrição da Doença por Jean-Martin Charcot. Charcot definiu os 4 sinais cardinais da Doença. Em 1817, Parkinson publicou em Londres um ensaio intitulado "An Essay on the Shaking Palsy", que vem a ser a primeira descrição mundial bem definida da DP. O ensaio definiu de forma geral a doença, determinou os sintomas principais, descreveu o diagnóstico diferencial com outras entidades e fez considerações a respeito da etiologia e também do tratamento. A enfermidade, intitulada "paralisia agitante", foi definida como doença caracterizada pela presença de movimentos involuntários tremulantes, com diminuição da força muscular, com tendência para a inclinação do tronco para frente e com alteração da marcha (festinação), tendo os sentidos e o intelecto não afetados. A evolução da doença foi caracterizada pela presença de tremores (principalmente das mãos e posteriormente mais difusos), com piora da marcha (passos curtos), quedas frequentes, obstipação, disartria, dificuldades para deglutição, sialorréia constante, incontinência urinária e finalmente anartria. Parkinson, na sua clássica descrição, ainda interrogou a possibilidade da medula espinhal cervical, na junção com a medula oblonga, ser a possível sede da doença, secundariamente a traumatismos locais. A doença somente tornou-se bem conhecida pelos neurologistas na segunda metade do século XIX. De todas as contribuições científicas no estudo da DP, após a descrição magistral de James Parkinson, sem sombra de dúvida a mais importante foi aquela realizada por Charcot. A contribuição de Charcot ao estudo da paralisia agitante foi de tamanha monta que seria muito justo que o seu nome tivesse sido acrescentado ao de Parkinson na nomeação da doença. Em primeiro lugar, foi Charcot que sugeriu a mudança do nome da enfermidade, de paralisia agitante para doença de Parkinson. Charcot acrescentou várias contribuições pessoais na descrição do quadro clínico, definindo a presença dos chamados quatro sinais cardinais da doença, quais sejam tremor, lentidão do movimento (bradicinesia), rigidez e dificuldades do equilíbrio, apresentando critérios para o diagnóstico diferencial e também sugerindo o primeiro tratamento para a doença (precursor dos alcalóides da beladona, hioscinamida). Contribuições de charcot: - Denominação doença de Parkinson - Caracterização do tremor de repouso (mãos) - Definição da rigidez muscular - Definição da instabilidade postural e da bradicinesia - Demonstração da ausência de fraqueza muscular - Caracterização da facies, postura, presença de disartria, disfagia, micrografia e alterações autonômicas Primeiro tratamento farmacológico
, tendo os sentidos e o intelecto não afetados. A evolução da doença foi caracterizada pela presença de tremores (principalmente das mãos e posteriormente mais difusos), com piora da marcha (passos curtos), quedas frequentes, obstipação, disartria, dificuldades para deglutição, sialorréia constante, incontinência urinária e finalmente anartria. Parkinson, na sua clássica descrição, ainda interrogou a possibilidade da medula espinhal cervical, na junção com a medula oblonga, ser a possível sede da doença, secundariamente a traumatismos locais. A doença somente tornou-se bem conhecida pelos neurologistas na segunda metade do século XIX. De todas as contribuições científicas no estudo da DP, após a descrição magistral de James Parkinson, sem sombra de dúvida a mais importante foi aquela realizada por Charcot. A contribuição de Charcot ao estudo da paralisia agitante foi de tamanha monta que seria muito justo que o seu nome tivesse sido acrescentado ao de Parkinson na nomeação da doença. Em primeiro lugar, foi Charcot que sugeriu a mudança do nome da enfermidade, de paralisia agitante para doença de Parkinson. Charcot acrescentou várias contribuições pessoais na descrição do quadro clínico, definindo a presença dos chamados quatro sinais cardinais da doença, quais sejam tremor, lentidão do movimento (bradicinesia), rigidez e dificuldades do equilíbrio, apresentando critérios para o diagnóstico diferencial e também sugerindo o primeiro tratamento para a doença (precursor dos alcalóides da beladona, hioscinamida). Contribuições de charcot: - Denominação doença de Parkinson. - Caracterização do tremor de repouso (mãos) - Definição da rigidez muscular. - Definição da instabilidade postural e da bradicinesia. - Demonstração da ausência de fraqueza muscular. - Caracterização da facies, postura, presença de disartria, disfagia, micrografia e. alterações autonômicas. Primeiro tratamento farmacológico.")
3
HISTÓRICO

4
HISTÓRICO precursor dos alcalóides da beladona
Contribuições de Charcot: - Denominação doença de Parkinson - Caracterização do tremor de repouso (mãos) - Definição da rigidez muscular - Definição da instabilidade postural e da bradicinesia - Demonstração da ausência de fraqueza muscular - Caracterização da facies, postura, presença de disartria, disfagia, micrografia e alterações autonômicas Primeiro tratamento farmacológico
 - Definição da rigidez muscular. - Definição da instabilidade postural e da bradicinesia. - Demonstração da ausência de fraqueza muscular. - Caracterização da facies, postura, presença de disartria, disfagia, micrografia e. alterações autonômicas. Primeiro tratamento farmacológico.")
5
EPIDEMIOLOGIA Segunda doença neurodegenerativa mais prevalente no mundo. Acomete 0.3% na população mundial, com incidência de 1-3% na população acima de 65 anos. 5 milhões de pacientes no mundo, com novos casos por habitantes/ano. Prevalência em homens (3:2). The male preponderance may be due to deficiency in a sex determining gene on the Y chromosome, the SRY gene, which is expressed in the substantia nigra only in males
. The male preponderance may be due to deficiency in a sex determining gene on the Y chromosome, the SRY gene, which is expressed in the substantia nigra only in males.")
6
EPIDEMIOLOGIA Fatores de proteção: - Tabaco - Cafeína - Atividade física moderada ou intensa - Uso de ibuprofeno Protective factors — An inverse correlation between PD and smoking is supported by the findings of large cohort studies and meta-analyses [86,88-90]. As an example, a 2012 meta-analysis reported that the risk of PD was significantly lower for current smokers compared with never smokers (relative risk [RR] 0.44, 95% CI ) [86]. In addition, the risk of PD was lower for ever smokers compared with never smokers (RR 0.64, 95% CI ). A neuroprotective effect of nicotine has been proposed as one possible explanation for these observations [91]. Despite some controversy, the evidence suggests that the reduced risk of PD associated with smoking should not simply be attributed to a "nonsmoking" personality in PD patients [93,94]. This alternative explanation posits that since dopamine is an integral component of the brain's reward system, people who will later develop signs of PD do not engage in reward-seeking behaviors, such as smoking, because dopamine is significantly depleted in the basal ganglia years before symptoms of PD appear [94]. One study showed that patients with PD had lower impulsive sensation seeking scores (and higher depression and anxiety scores) [95]. However, impulse control disorders and compulsive behavior become markedly increased with dopaminergic therapy, particularly with the dopamine agonists. Data from several prospective studies suggest that moderate to vigorous physical activity and exercise is associated with a reduced risk for developing PD [98-102]. However, an alternative explanation for the association is reversed causality, given that reduced physical activity may be a preclinical sign of PD. There is evidence from several meta-analyses that ibuprofen may be associated with a reduced risk of PD [ ]. The data regarding other nonsteroidal antiinflammatory drugs (NSAIDs) are conflicting, with some meta-analyses finding that NSAIDs are associated with a reduced risk of PD [86,104], and others finding no significant association [103,105,106].
 [86]. In addition, the risk of PD was lower for ever smokers compared with never smokers (RR 0.64, 95% CI ). A neuroprotective effect of nicotine has been proposed as one possible explanation for these observations [91]. Despite some controversy, the evidence suggests that the reduced risk of PD associated with smoking should not simply be attributed to a nonsmoking personality in PD patients [93,94]. This alternative explanation posits that since dopamine is an integral component of the brain s reward system, people who will later develop signs of PD do not engage in reward-seeking behaviors, such as smoking, because dopamine is significantly depleted in the basal ganglia years before symptoms of PD appear [94]. One study showed that patients with PD had lower impulsive sensation seeking scores (and higher depression and anxiety scores) [95]. However, impulse control disorders and compulsive behavior become markedly increased with dopaminergic therapy, particularly with the dopamine agonists. Data from several prospective studies suggest that moderate to vigorous physical activity and exercise is associated with a reduced risk for developing PD [98-102]. However, an alternative explanation for the association is reversed causality, given that reduced physical activity may be a preclinical sign of PD. There is evidence from several meta-analyses that ibuprofen may be associated with a reduced risk of PD [ ]. The data regarding other nonsteroidal antiinflammatory drugs (NSAIDs) are conflicting, with some meta-analyses finding that NSAIDs are associated with a reduced risk of PD [86,104], and others finding no significant association [103,105,106].")
7
EPIDEMIOLOGIA Fatores de Risco: - História Familiar de Doença de Parkinson - Exposição à Agrotóxicos Fatores de risco comprovados, com nível de evidência A.

8
EPIDEMIOLOGIA Outros Fatores de Risco: - Histórico de TCE - Viver em áreas urbanas ou industriais em que haja liberação de Mn, Cu ou Pb - Exposição à Solventes, principalmente tricloroetileno - Viver em área rural - Trabalho agrícola - Beber água de poço - Consumo de leite - Excesso de peso - Dieta rica em ferro e manganês - Alto nível educacional - Histórico de anemia Fatores de risco cujas evidências são inconclusivas.

9
PARKINSONISMO “Síndrome clínica caracterizada pela presença de qualquer combinação de 2 dos 4 sinais cardinais: bradicinesia, rigidez muscular, tremor de repouso e instabilidade postural” 80% das síndromes parkinsonianas são provocadas pela doença de parkinson esporádica ou familiar. Tremor de repouso de 4 a 5Hz.

10
ETIOLOGIAS Parkinsonismo Primário – Doença de Parkinson esporádica ou familiar. Parkinsonismo Secundário – Infeccioso, Metabólico, Trauma, Tumor, Hipóxia, Intoxicação. Síndromes Parkinson-Plus – Degeneração ganglionar, Síndromes de demência, Lytico-Bodig. Doenças Heredodegenerativas – Wilson, Hallervorden-Spatz, Lubag, Neuroacantocitose. Mencionar que a tabela mostrando todas as possíveis causas da síndrome parkinsoniana é gigantesca. Enfatizar que o restante da aula irá falar apenas sobre a A Doença de Parkinson esporádica ou familiar.

12
FISIOPATOLOGIA A patologia bioquímica central na Doença de Parkinson é a diminuição da neurotransmissão Dopaminérgica nos GB. A depleção de Dopamina ocorre devido à despigmentação, gliose e morte neuronal na SNc. PATHOLOGY — Depigmentation, neuronal loss, and gliosis, particularly in the substantia nigra pars compacta and in the pontine locus ceruleus, are typical abnormalities found in the brains of patients with PD. Neuronal degeneration is also present in the dorsal nucleus of the vagus in the medulla and other brainstem nuclei. # a patologia bioquímica central no parkinsonismo é a diminuição da neurotransmissão dopaminérgica nos GB. #Isso ocorre devido à despigmentação, gliose e morte neuronal na SNc. Esse fenômeno também ocorre no Locus ceruleus e nos núcleos dorsais do vago. # a porção ventrolateral da SN, a qual se projeta para o putamen, é a mais atingida na doença. Em contraste, no envelhecimento normal, a primeira porção a se degenerar é a dorsal, provocando depleção de dopamina no núcleo caudado.

13
FISIOPATOLOGIA This schematic drawing of the basal ganglia and their connections illustrates the main striato-fugal pathways. In Parkinson disease (PD), the indirect inhibitory pathway mediated via D2 striatal receptors is postulated to be overactive, whereas the direct inhibitory pathway mediated via D1 striatal receptors is underactive. As a result, the main pallidal-thalamic outflow pathway provides excessive inhibitory input to the thalamus, which causes suppression of thalamo-cortical-spinal pathway, manifested clinically by paucity and slowness of movement (bradykinesia). Green: excitatory; Red: inhibitory; GPe: globus pallidus externa; GPi: globus pallidus interna; SNc: substantia nigra pars compacta; SNr: substantia nigra pars reticulata; STN: subthalamic nucleus; TH: ventrolateral nucleus of the thalamus. Because of the apparent discrepancy between loss of striatal dopamine (>80 percent) and the degree of loss of neurons in the substantia nigra (50 to 60 percent), some have suggested that the initial site of pathology is in the striatum and that retrograde degeneration may be responsible for the neuronal loss in the substantia nigra [3]. An alternative explanation is that each dopaminergic neuron has multiple projections that terminate in the striatum, so that death of the cell body has a multiplying effect on loss of terminals. Five distinct dopamine receptors (D1 through D5) have been cloned and characterized; they are found throughout the basal ganglia and limbic system. The D1 and D2 receptors are highly concentrated in the dorsal (motor) striatum and are the most relevant to the pathophysiology of PD because they are activated by the dopaminergic pathway originating in the SNc and terminating in the caudate and putamen. Receptors designated as D3, D4, and D5 are more abundant in the mesolimbic or emotional part of the brain (D3, D4) and hippocampus/hypothalamus (D5) [5]. ●The indirect pathway is mediated chiefly via dopamine's inhibitory influence on striatal D2 dopamine receptors. In the indirect pathway, the striatum projects to the neurons in the lateral GP (GPe) utilizing GABA, and the GPe in turn projects to the STN, which provides excitatory input via glutamate to the internal segment of the GP (GPi) and SN pars reticulata (SNr). GPi neurons are GABAergic and synapse in the ventrolateral nucleus of the thalamus. Thalamic input to the cortex is excitatory.
, the indirect inhibitory pathway mediated via D2 striatal receptors is postulated to be overactive, whereas the direct inhibitory pathway mediated via D1 striatal receptors is underactive. As a result, the main pallidal-thalamic outflow pathway provides excessive inhibitory input to the thalamus, which causes suppression of thalamo-cortical-spinal pathway, manifested clinically by paucity and slowness of movement (bradykinesia). Green: excitatory; Red: inhibitory; GPe: globus pallidus externa; GPi: globus pallidus interna; SNc: substantia nigra pars compacta; SNr: substantia nigra pars reticulata; STN: subthalamic nucleus; TH: ventrolateral nucleus of the thalamus. Because of the apparent discrepancy between loss of striatal dopamine (>80 percent) and the degree of loss of neurons in the substantia nigra (50 to 60 percent), some have suggested that the initial site of pathology is in the striatum and that retrograde degeneration may be responsible for the neuronal loss in the substantia nigra [3]. An alternative explanation is that each dopaminergic neuron has multiple projections that terminate in the striatum, so that death of the cell body has a multiplying effect on loss of terminals. Five distinct dopamine receptors (D1 through D5) have been cloned and characterized; they are found throughout the basal ganglia and limbic system. The D1 and D2 receptors are highly concentrated in the dorsal (motor) striatum and are the most relevant to the pathophysiology of PD because they are activated by the dopaminergic pathway originating in the SNc and terminating in the caudate and putamen. Receptors designated as D3, D4, and D5 are more abundant in the mesolimbic or emotional part of the brain (D3, D4) and hippocampus/hypothalamus (D5) [5]. ●The indirect pathway is mediated chiefly via dopamine s inhibitory influence on striatal D2 dopamine receptors. In the indirect pathway, the striatum projects to the neurons in the lateral GP (GPe) utilizing GABA, and the GPe in turn projects to the STN, which provides excitatory input via glutamate to the internal segment of the GP (GPi) and SN pars reticulata (SNr). GPi neurons are GABAergic and synapse in the ventrolateral nucleus of the thalamus. Thalamic input to the cortex is excitatory.")
14
FISIOPATOLOGIA This schematic drawing of the basal ganglia and their connections illustrates the main striato-fugal pathways. In Parkinson disease (PD), the indirect inhibitory pathway mediated via D2 striatal receptors is postulated to be overactive, whereas the direct inhibitory pathway mediated via D1 striatal receptors is underactive. As a result, the main pallidal-thalamic outflow pathway provides excessive inhibitory input to the thalamus, which causes suppression of thalamo-cortical-spinal pathway, manifested clinically by paucity and slowness of movement (bradykinesia). Green: excitatory; Red: inhibitory; GPe: globus pallidus externa; GPi: globus pallidus interna; SNc: substantia nigra pars compacta; SNr: substantia nigra pars reticulata; STN: subthalamic nucleus; TH: ventrolateral nucleus of the thalamus. Because of the apparent discrepancy between loss of striatal dopamine (>80 percent) and the degree of loss of neurons in the substantia nigra (50 to 60 percent), some have suggested that the initial site of pathology is in the striatum and that retrograde degeneration may be responsible for the neuronal loss in the substantia nigra [3]. An alternative explanation is that each dopaminergic neuron has multiple projections that terminate in the striatum, so that death of the cell body has a multiplying effect on loss of terminals. Five distinct dopamine receptors (D1 through D5) have been cloned and characterized; they are found throughout the basal ganglia and limbic system. The D1 and D2 receptors are highly concentrated in the dorsal (motor) striatum and are the most relevant to the pathophysiology of PD because they are activated by the dopaminergic pathway originating in the SNc and terminating in the caudate and putamen. Receptors designated as D3, D4, and D5 are more abundant in the mesolimbic or emotional part of the brain (D3, D4) and hippocampus/hypothalamus (D5) [5]. ●The indirect pathway is mediated chiefly via dopamine's inhibitory influence on striatal D2 dopamine receptors. In the indirect pathway, the striatum projects to the neurons in the lateral GP (GPe) utilizing GABA, and the GPe in turn projects to the STN, which provides excitatory input via glutamate to the internal segment of the GP (GPi) and SN pars reticulata (SNr). GPi neurons are GABAergic and synapse in the ventrolateral nucleus of the thalamus. Thalamic input to the cortex is excitatory.
, the indirect inhibitory pathway mediated via D2 striatal receptors is postulated to be overactive, whereas the direct inhibitory pathway mediated via D1 striatal receptors is underactive. As a result, the main pallidal-thalamic outflow pathway provides excessive inhibitory input to the thalamus, which causes suppression of thalamo-cortical-spinal pathway, manifested clinically by paucity and slowness of movement (bradykinesia). Green: excitatory; Red: inhibitory; GPe: globus pallidus externa; GPi: globus pallidus interna; SNc: substantia nigra pars compacta; SNr: substantia nigra pars reticulata; STN: subthalamic nucleus; TH: ventrolateral nucleus of the thalamus. Because of the apparent discrepancy between loss of striatal dopamine (>80 percent) and the degree of loss of neurons in the substantia nigra (50 to 60 percent), some have suggested that the initial site of pathology is in the striatum and that retrograde degeneration may be responsible for the neuronal loss in the substantia nigra [3]. An alternative explanation is that each dopaminergic neuron has multiple projections that terminate in the striatum, so that death of the cell body has a multiplying effect on loss of terminals. Five distinct dopamine receptors (D1 through D5) have been cloned and characterized; they are found throughout the basal ganglia and limbic system. The D1 and D2 receptors are highly concentrated in the dorsal (motor) striatum and are the most relevant to the pathophysiology of PD because they are activated by the dopaminergic pathway originating in the SNc and terminating in the caudate and putamen. Receptors designated as D3, D4, and D5 are more abundant in the mesolimbic or emotional part of the brain (D3, D4) and hippocampus/hypothalamus (D5) [5]. ●The indirect pathway is mediated chiefly via dopamine s inhibitory influence on striatal D2 dopamine receptors. In the indirect pathway, the striatum projects to the neurons in the lateral GP (GPe) utilizing GABA, and the GPe in turn projects to the STN, which provides excitatory input via glutamate to the internal segment of the GP (GPi) and SN pars reticulata (SNr). GPi neurons are GABAergic and synapse in the ventrolateral nucleus of the thalamus. Thalamic input to the cortex is excitatory.")
15
FISIOPATOLOGIA Ao aparecimento dos sintomas, estima-se que houve perda de 60% dos neurônios dopaminérgicos da SNc. Diversos mecanismos de compensação atuam na fase pré-sintomática da doença. Using a quantitative method, one study of seven patients with PD and seven controls found that the number of pigmented neurons in the substantia nigra, normally 550,000, was reduced by 66 percent in those with PD [16]. In addition, the number of nonpigmented neurons, normally 260,000, was reduced by 24 percent. By the time the first symptoms of PD emerge, about 60 percent of the neurons in the substantia nigra pars compact have been lost [3]. # o número de neurônios pigmentados na substância negra gira em torno de Esse número é 66% menor em pacientes com doença de Parkinson. Em relação aos neurônios não pigmentados, o número é 24% menor em pacientes com PD em relação aos controles.

16
FISIOPATOLOGIA MECANISMOS DE COMPENSAÇÃO
Aumento da síntese de Dopamina nos neurônios sobreviventes. Proliferação de receptores D2. Aumento da número de aferentes dendríticos. Junções GAP entre neurônios striatais. Redução da recaptação de Dopamina. Compensatory mechanisms # increasing the synthesis of dopamine in surviving neurons # increasing the afferents to the dendrites of dopaminergic neurons. #proliferation of D2 receptors. # gap junctions, which allow rapid communications between striatal neurons, increase dramatically after dopaminergic denervation [11]. #downregulation of the dopamine transporter, resulting in less dopamine reuptake and higher synaptic dopamine levels [15]. Three stages of compensation during the presymptomatic period of PD have been proposed [14]: ●An early period during which the dopamine homeostatic compensatory mechanisms discussed above are capable of "masking" the disease ●Increased activity of the basal ganglia output nuclei (eg, internal segment of the globus pallidus) as striatal dopamine homeostasis breaks down ●Increased intensity of compensation in structures outside of the basal ganglia (eg, supplementary motor area of the cortex) as parkinsonian motor abnormalities emerge.
 as striatal dopamine homeostasis breaks down. ●Increased intensity of compensation in structures outside of the basal ganglia (eg, supplementary motor area of the cortex) as parkinsonian motor abnormalities emerge.")
17
FISIOPATOLOGIA # Independente do gatilho inicial, a neurodegeneração envolve mecanismos de necrose e apoptose. Protein misfolding, aggregation, and toxicity — Mutations in the gene on chromosome 4q21.3-q22 that codes for alpha-synuclein have emerged as one of the most important elements of cell death in various neurodegenerative disorders, together known as synucleinopathies [40]. Defective proteolysis — Cellular protein homeostasis is normally maintained primarily by three coordinated pathways – molecular chaperones, the ubiquitin-proteasome system, and the autophagy-lysosomal pathway – that mediate the repair or removal of abnormal proteins [50-52]. While the data are not entirely consistent, it appears that all three pathways are involved in the processing of alpha-synuclein. When these systems are inhibited or impaired, abnormal proteins such as mutated alpha-synuclein can misfold, aggregate, and clog the normal molecular traffic of the cell, leading to cell death. Mitochondrial dysfunction — The role of mitochondria in the pathogenesis of PD was first suggested by discovery of the association between the meperidine analogue 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and parkinsonism [55,56]. The oxidation of MPTP produces 1-methyl-4-phenylpyridium (MPP+), which is taken up by dopaminergic terminals, selectively inhibits mitochondrial complex I activity, disrupts calcium homeostasis, and induces endoplasmic reticulum stress, resulting in cell damage [56,57]. ●A cellular insult (eg, oxidative stress, excitotoxicity, DNA damage) increases cytosolic calcium and oxidative radicals, and activates nuclear enzyme poly(ADP-ribose) polymerase-1 (PARP-1) leading to the formation of poly(ADP-ribose PAR) ●This leads to decreased mitochondrial membrane potential, which in turn opens mitochondrial permeability transition pores (PTP) ●Release of NAD+ through the PTP leads to NAD+ depletion ●Release of mitochondrial apoptosis initiating factors promotes release of cytochrome c, which leads to activation of the "executioner" enzyme caspase and to apoptosis Oxidative stress — The oxidative stress hypothesis postulates that inappropriate production of reactive oxygen species leads to neurodegeneration [59,66]. Dopamine is normally metabolized not only by monoamine oxidase-mediated enzymatic oxidation, but also by auto-oxidation to neuromelanin. Iron metabolism — Elemental iron plays a critical role in oxidative metabolism and it also serves as a cofactor in the synthesis of neurotransmitters [69]. It is increased by about 50 percent in substantia nigra of PD brains relative to controls [70], suggesting that abnormal iron metabolism plays a pathologic role in the development of PD [71]. The role of inflammatory processes in the pathogenesis of PD is further supported by the following observations: ●Cyclooxygenase-2, the rate-limiting enzyme in prostaglandin E2 synthesis, appears to be upregulated in patients with PD and in the MPTP mouse model of PD; cyclooxygenase-2 inhibition prevents the formation of potentially toxic dopamine-quinones in MPTP mice and presumably in patients with PD [76]. ●In a positron emission tomography study that used markers for activated microglia and for dopamine transporter, microglial activity in patients with PD correlated with decreased density of dopamine transporter [77]. ●Infiltration of CD4+ T lymphocytes contributed to neuronal cell death in a mouse model of PD
 and parkinsonism [55,56]. The oxidation of MPTP produces 1-methyl-4-phenylpyridium (MPP+), which is taken up by dopaminergic terminals, selectively inhibits mitochondrial complex I activity, disrupts calcium homeostasis, and induces endoplasmic reticulum stress, resulting in cell damage [56,57]. ●A cellular insult (eg, oxidative stress, excitotoxicity, DNA damage) increases cytosolic calcium and oxidative radicals, and activates nuclear enzyme poly(ADP-ribose) polymerase-1 (PARP-1) leading to the formation of poly(ADP-ribose PAR) ●This leads to decreased mitochondrial membrane potential, which in turn opens mitochondrial permeability transition pores (PTP) ●Release of NAD+ through the PTP leads to NAD+ depletion. ●Release of mitochondrial apoptosis initiating factors promotes release of cytochrome c, which leads to activation of the executioner enzyme caspase and to apoptosis. Oxidative stress — The oxidative stress hypothesis postulates that inappropriate production of reactive oxygen species leads to neurodegeneration [59,66]. Dopamine is normally metabolized not only by monoamine oxidase-mediated enzymatic oxidation, but also by auto-oxidation to neuromelanin. Iron metabolism — Elemental iron plays a critical role in oxidative metabolism and it also serves as a cofactor in the synthesis of neurotransmitters [69]. It is increased by about 50 percent in substantia nigra of PD brains relative to controls [70], suggesting that abnormal iron metabolism plays a pathologic role in the development of PD [71]. The role of inflammatory processes in the pathogenesis of PD is further supported by the following observations: ●Cyclooxygenase-2, the rate-limiting enzyme in prostaglandin E2 synthesis, appears to be upregulated in patients with PD and in the MPTP mouse model of PD; cyclooxygenase-2 inhibition prevents the formation of potentially toxic dopamine-quinones in MPTP mice and presumably in patients with PD [76]. ●In a positron emission tomography study that used markers for activated microglia and for dopamine transporter, microglial activity in patients with PD correlated with decreased density of dopamine transporter [77]. ●Infiltration of CD4+ T lymphocytes contributed to neuronal cell death in a mouse model of PD.")
18
FISIOPATOLOGIA

19
FISIOPATOLOGIA Não existe consenso a respeito dos critérios para o Diagnóstico Patológico da Doença de Parkinson. Corpos de Lewy são considerados o “marco” patológico da DP. # Não existe consenso a respeito dos critérios para o diagnóstico patológico de PD. # Corpos de Lewy são inclusões eosinofílicas intracitoplasmáticas nos neurônios, compostas principalmente de alfa-sinucleína e ubiquitina arranjadas de forma fibrilar. São considerados o “marco” patológico da PD. #Lewy bodies are seen in the substantia nigra, the basal nucleus of Meynert, locus ceruleus, cerebral cortex, sympathetic ganglia, the dorsal vagal nucleus, the myenteric plexus of the intestines, and even in the cardiac sympathetic plexus. #Lewy bodies are not specific for PD, since they are found in as many as 10 percent of brains of normal elderly persons, and in patients with other neurodegenerative diseases Inclusions such as Lewy bodies have traditionally been considered toxic. However, some studies suggest that they may actually be neuroprotective, and that compounds that promote the formation of inclusions lessen the pathology of PD.

20
BRAAK STAGING Braak staging — In the traditional view, the pathologic process of PD starts with degeneration of dopaminergic neurons in the substantia nigra. This view has been challenged by the neuropathologist Heiko Braak, has proposed that the pathologic changes of PD start in the medulla of the brainstem and in the olfactory bulb, progressing rostrally over many years to the cerebral cortex in a predictable six-stage process (figure 2) [26,27]. According to Braak staging, the progression of pathologic changes occurs as follows [26]: ●During presymptomatic stages 1 and 2, the pathologic changes are found in the medulla oblongata and olfactory bulb. ●In stages 3 and 4, the pathology has migrated rostrally to the substantia nigra pars compacta and other neuronal clusters of the midbrain and basal forebrain, at which time the classic motor symptoms of PD first appear. ●In end-stages 5 and 6, the pathologic process encroaches upon the telencephalic cortex of the temporal and frontal lobes. However, the validity and predictive utility of Braak's staging has been questioned, as there are no cell counts to correlate with the described synuclein pathology and no observed asymmetry in the pathologic findings that correlate with the well-recognized asymmetry of clinical findings [28,29].
 [26,27]. According to Braak staging, the progression of pathologic changes occurs as follows [26]: ●During presymptomatic stages 1 and 2, the pathologic changes are found in the medulla oblongata and olfactory bulb. ●In stages 3 and 4, the pathology has migrated rostrally to the substantia nigra pars compacta and other neuronal clusters of the midbrain and basal forebrain, at which time the classic motor symptoms of PD first appear. ●In end-stages 5 and 6, the pathologic process encroaches upon the telencephalic cortex of the temporal and frontal lobes. However, the validity and predictive utility of Braak s staging has been questioned, as there are no cell counts to correlate with the described synuclein pathology and no observed asymmetry in the pathologic findings that correlate with the well-recognized asymmetry of clinical findings [28,29].")
21
GENÉTICA

22
SINAIS E SINTOMAS Tremor Bradicinesia Rigidez Instabilidade Postural
Tremor — The tremor in PD, typically described as "pill-rolling", is a rest tremor, meaning that it is most noticeable when the tremulous body part is supported by gravity and not engaged in purposeful activities. Tremors in other conditions, such as essential tremor or multiple sclerosis, are typically action tremors, in which the tremor occurs when the affected limb is being used. (See "Overview of tremor".) It is not unusual for a parkinsonian tremor to be present with postural maneuvers or with action, but in such cases, the tremor is typically much more severe at rest. Because parkinsonian tremor decreases with purposeful action, it is usually the least disabling of the cardinal manifestations. However, when the tremor is severe, it can be difficult to distinguish a primary resting tremor from a primary action tremor. Some patients with PD may have a re-emergent tremor: a postural tremor that manifests after a latency of several seconds and has a frequency typical of the rest tremor in PD [11,12]. This distinction is important, as patients with PD who have a re-emergent postural tremor may be misdiagnosed as having essential tremor [13]. The tremor of early PD is most often intermittent, and may not be noticeable to others. In fact, about half of patients with PD report a sensation of internal tremulousness in the limbs or body that is unrelated to the presence of observable tremor [14]. However, as the disease progresses, tremor usually becomes evident. In the clinical setting, tremor in the limbs can be seen when the patient is relaxed with the hands resting quietly on the lap. Distracting the patient by asking him or her to perform mental calculations or voluntary repetitive movements of the contralateral limb often accentuates a mild tremor and may uncover a latent tremor. A resting hand tremor may be present only during the gait evaluation. The frequency of the tremor in PD is between 3 and 7 Hz, and most often is between 4 and 5 Hz [15]. Tremor is the presenting symptom in approximately 70 percent of patients with PD [16], and the percentage of patients with tremor at some point in the course of the disease is high, ranging from 79 to 100 percent [16-20]. Tremor usually starts unilaterally in the hand, and then spreads contralaterally several years after the onset of symptoms [21]. The side that is initially affected tends to be the more affected side throughout the course of the disease. The tremor of PD can also involve the legs, lips, jaw, and tongue, but rarely involves the head [15,22]. Anxiety, emotional excitement, or stressful situations can exacerbate the tremor. Bradykinesia — Bradykinesia is a generalized slowness of movement. It is arguably the major cause of disability in PD and is eventually seen in almost all patients. While it is the most common feature in PD, it is also the most difficult symptom for patients to describe. "Weakness," "incoordination," and "tiredness" are often used to describe the decreased ability to initiate voluntary movement. In the arms, bradykinesia typically starts distally with decreased manual dexterity of the fingers. Patients often complain of difficulty performing simple tasks, such as buttoning clothes, tying shoelaces, double clicking a computer mouse, typing, or lifting coins from a pocket or purse. In the legs, common complaints related to bradykinesia when walking include dragging the legs, shorter (shuffling) steps, or a feeling of unsteadiness. Patients may also have difficulty standing up from a chair or getting out of a car. As the disease progresses, gait freezing and festination may develop. James Parkinson defined festination as "an irresistible impulse to take much quicker and shorter steps, and thereby to adopt unwillingly a running pace" [1]. Clinical examination of bradykinesia includes evaluation of limb movements on each side of the body. The speed, amplitude, and rhythm of finger tapping, hand gripping, pronation-supination hand movements, and heel or toe tapping should be carefully observed. In mild PD, these tasks usually show some slowing and decreased amplitude when observed for more than a few seconds. As the disease progresses movements become less coordinated, with frequent hesitations or arrests. Rigidity — Rigidity is an increased resistance to passive movement about a joint and occurs in approximately 90 percent of patients with PD [16-19]. Rigidity, like tremor and bradykinesia, often begins unilaterally, and typically on the same side as the tremor if one is present. Rigidity eventually progresses to the contralateral side, and remains asymmetric throughout the disease [21]. Cogwheel rigidity can be seen in PD, and the term refers to a ratchety pattern of resistance and relaxation as the examiner moves the limb through its full range of motion [23]. This phenomenon is thought to be a manifestation of tremor superimposed on increased tone [24]. However, not all patients with PD have cogwheel rigidity; many instead will have lead-pipe rigidity, a tonic resistance that is smooth throughout the entire range of passive movement. Rigidity can affect any part of the body, and may contribute to complaints of stiffness and pain. Features, such as the striatal hand (extension of the proximal and distal interphalangeal joints with flexion at the metacarpophalangeal joints), decreased arm swing with walking, and the typical stooped posture, result, at least in part, from rigidity. At the bedside, rigidity is tested by passively manipulating the limbs. It can be brought out by having the patient perform repetitive maneuvers using the contralateral limb or by performing mental arithmetic. Postural instability — Postural instability is an impairment of centrally-mediated postural reflexes that cause a feeling of imbalance and a tendency to fall with a significant risk of injury. Because postural instability usually does not appear until later in the course of PD, patients with parkinsonian signs who fall early in the course of the illness most likely have another parkinsonian syndrome, such as progressive supranuclear palsy or multiple system atrophy. (See "Diagnosis of Parkinson disease", section on 'Differential diagnosis'.) Postural instability is tested clinically with the "pull" test, where the examiner stands behind the patient and firmly pulls the patient by the shoulders. Patients with normal postural reflexes should be able to maintain balance and retropulse (step backward) no more than one step. Patients with PD and postural instability, on the other hand, are likely to fall or take multiple steps backwards. Initially, a positive pull test may be the only sign of balance impairment. However, as postural instability progresses, the gait may show signs of festination. Once postural reflexes are lost, patients are generally wheelchair-bound. Among the primary motor features of PD, postural instability is the least responsive to dopaminergic therapies [25]. In addition, postural instability and gait difficulty are major contributors to disability in patients with PD
 It is not unusual for a parkinsonian tremor to be present with postural maneuvers or with action, but in such cases, the tremor is typically much more severe at rest. Because parkinsonian tremor decreases with purposeful action, it is usually the least disabling of the cardinal manifestations. However, when the tremor is severe, it can be difficult to distinguish a primary resting tremor from a primary action tremor. Some patients with PD may have a re-emergent tremor: a postural tremor that manifests after a latency of several seconds and has a frequency typical of the rest tremor in PD [11,12]. This distinction is important, as patients with PD who have a re-emergent postural tremor may be misdiagnosed as having essential tremor [13]. The tremor of early PD is most often intermittent, and may not be noticeable to others. In fact, about half of patients with PD report a sensation of internal tremulousness in the limbs or body that is unrelated to the presence of observable tremor [14]. However, as the disease progresses, tremor usually becomes evident. In the clinical setting, tremor in the limbs can be seen when the patient is relaxed with the hands resting quietly on the lap. Distracting the patient by asking him or her to perform mental calculations or voluntary repetitive movements of the contralateral limb often accentuates a mild tremor and may uncover a latent tremor. A resting hand tremor may be present only during the gait evaluation. The frequency of the tremor in PD is between 3 and 7 Hz, and most often is between 4 and 5 Hz [15]. Tremor is the presenting symptom in approximately 70 percent of patients with PD [16], and the percentage of patients with tremor at some point in the course of the disease is high, ranging from 79 to 100 percent [16-20]. Tremor usually starts unilaterally in the hand, and then spreads contralaterally several years after the onset of symptoms [21]. The side that is initially affected tends to be the more affected side throughout the course of the disease. The tremor of PD can also involve the legs, lips, jaw, and tongue, but rarely involves the head [15,22]. Anxiety, emotional excitement, or stressful situations can exacerbate the tremor. Bradykinesia — Bradykinesia is a generalized slowness of movement. It is arguably the major cause of disability in PD and is eventually seen in almost all patients. While it is the most common feature in PD, it is also the most difficult symptom for patients to describe. Weakness, incoordination, and tiredness are often used to describe the decreased ability to initiate voluntary movement. In the arms, bradykinesia typically starts distally with decreased manual dexterity of the fingers. Patients often complain of difficulty performing simple tasks, such as buttoning clothes, tying shoelaces, double clicking a computer mouse, typing, or lifting coins from a pocket or purse. In the legs, common complaints related to bradykinesia when walking include dragging the legs, shorter (shuffling) steps, or a feeling of unsteadiness. Patients may also have difficulty standing up from a chair or getting out of a car. As the disease progresses, gait freezing and festination may develop. James Parkinson defined festination as an irresistible impulse to take much quicker and shorter steps, and thereby to adopt unwillingly a running pace [1]. Clinical examination of bradykinesia includes evaluation of limb movements on each side of the body. The speed, amplitude, and rhythm of finger tapping, hand gripping, pronation-supination hand movements, and heel or toe tapping should be carefully observed. In mild PD, these tasks usually show some slowing and decreased amplitude when observed for more than a few seconds. As the disease progresses movements become less coordinated, with frequent hesitations or arrests. Rigidity — Rigidity is an increased resistance to passive movement about a joint and occurs in approximately 90 percent of patients with PD [16-19]. Rigidity, like tremor and bradykinesia, often begins unilaterally, and typically on the same side as the tremor if one is present. Rigidity eventually progresses to the contralateral side, and remains asymmetric throughout the disease [21]. Cogwheel rigidity can be seen in PD, and the term refers to a ratchety pattern of resistance and relaxation as the examiner moves the limb through its full range of motion [23]. This phenomenon is thought to be a manifestation of tremor superimposed on increased tone [24]. However, not all patients with PD have cogwheel rigidity; many instead will have lead-pipe rigidity, a tonic resistance that is smooth throughout the entire range of passive movement. Rigidity can affect any part of the body, and may contribute to complaints of stiffness and pain. Features, such as the striatal hand (extension of the proximal and distal interphalangeal joints with flexion at the metacarpophalangeal joints), decreased arm swing with walking, and the typical stooped posture, result, at least in part, from rigidity. At the bedside, rigidity is tested by passively manipulating the limbs. It can be brought out by having the patient perform repetitive maneuvers using the contralateral limb or by performing mental arithmetic. Postural instability — Postural instability is an impairment of centrally-mediated postural reflexes that cause a feeling of imbalance and a tendency to fall with a significant risk of injury. Because postural instability usually does not appear until later in the course of PD, patients with parkinsonian signs who fall early in the course of the illness most likely have another parkinsonian syndrome, such as progressive supranuclear palsy or multiple system atrophy. (See Diagnosis of Parkinson disease , section on Differential diagnosis .) Postural instability is tested clinically with the pull test, where the examiner stands behind the patient and firmly pulls the patient by the shoulders. Patients with normal postural reflexes should be able to maintain balance and retropulse (step backward) no more than one step. Patients with PD and postural instability, on the other hand, are likely to fall or take multiple steps backwards. Initially, a positive pull test may be the only sign of balance impairment. However, as postural instability progresses, the gait may show signs of festination. Once postural reflexes are lost, patients are generally wheelchair-bound. Among the primary motor features of PD, postural instability is the least responsive to dopaminergic therapies [25]. In addition, postural instability and gait difficulty are major contributors to disability in patients with PD.")
23
MARCHA PARKINSONIANA

24
“PILL-ROLLING” TREMOR

25
SINAIS E SINTOMAS Outros sintomas incluem: - Hipomimia - Disartria hipocinética - Disfagia - Micrografia - Sialorréia - Visão embaçada - Festinação - Diminuição no número de piscadas OTHER MOTOR FEATURES — In addition to the cardinal manifestations just discussed, there are a number of other motor features seen in Parkinson disease (PD) (table 1): ●Craniofacial •Hypomimia (masked facial expression) •Decreased spontaneous eye blink rate •Speech impairment, including hypokinetic dysarthria, hypophonia, and palilalia (repetition of a phrase or word with increasing rapidity) •Dysphagia •Sialorrhea ●Visual •Blurred vision •Impaired contrast sensitivity •Hypometric saccades •Impaired vestibuloocular reflex •Impaired upward gaze and convergence •Eyelid opening apraxia ●Musculoskeletal •Micrographia •Dystonia •Myoclonus •Stooped posture •Camptocormia (severe anterior flexion of the thoracolumbar spine) •Kyphosis •Scoliosis •Difficulty turning in bed ●Gait •Shuffling, short-stepped gait •Freezing •Festination
 (table 1): ●Craniofacial. •Hypomimia (masked facial expression) •Decreased spontaneous eye blink rate. •Speech impairment, including hypokinetic dysarthria, hypophonia, and palilalia (repetition of a phrase or word with increasing rapidity) •Dysphagia. •Sialorrhea. ●Visual. •Blurred vision. •Impaired contrast sensitivity. •Hypometric saccades. •Impaired vestibuloocular reflex. •Impaired upward gaze and convergence. •Eyelid opening apraxia. ●Musculoskeletal. •Micrographia. •Dystonia. •Myoclonus. •Stooped posture. •Camptocormia (severe anterior flexion of the thoracolumbar spine) •Kyphosis. •Scoliosis. •Difficulty turning in bed. ●Gait. •Shuffling, short-stepped gait. •Freezing. •Festination.")
26
FISIOPATOLOGIA NONMOTOR SYMPTOMS — Parkinson disease (PD) has traditionally been considered a motor system disorder, but it is now widely recognized to be a complex disorder with diverse clinical features that include neuropsychiatric and nonmotor manifestations in addition to its motor symptomatology [33-36]. These features include the following: ●Cognitive dysfunction and dementia ●Psychosis and hallucinations ●Mood disorders including depression, anxiety, and apathy/abulia ●Sleep disturbances ●Fatigue ●Autonomic dysfunction ●Olfactory dysfunction ●Pain and sensory disturbances ●Dermatologic findings (seborrhea) ●Rhinorrhea In a multicenter survey of over 1000 patients with PD, virtually all (97 percent) patients reported nonmotor symptoms, with each patient experiencing an average of approximately eight nonmotor symptoms [34]. Nonmotor symptoms in the psychiatric domain occurred most frequently. Psychiatric symptoms such as psychosis or dementia may cause more disability than the motor features and may be more difficult to treat. In a single center survey of 265 patients with PD, pain, mood disorders, and sleep problems were the most troublesome nonmotor symptoms occurring in both early and late stage PD [37]. In some patients, certain nonmotor features of PD (eg, olfactory dysfunction, constipation, depression, and REM sleep behavior disorder) may present before the motor ones [5].
 has traditionally been considered a motor system disorder, but it is now widely recognized to be a complex disorder with diverse clinical features that include neuropsychiatric and nonmotor manifestations in addition to its motor symptomatology [33-36]. These features include the following: ●Cognitive dysfunction and dementia. ●Psychosis and hallucinations. ●Mood disorders including depression, anxiety, and apathy/abulia. ●Sleep disturbances. ●Fatigue. ●Autonomic dysfunction. ●Olfactory dysfunction. ●Pain and sensory disturbances. ●Dermatologic findings (seborrhea) ●Rhinorrhea. In a multicenter survey of over 1000 patients with PD, virtually all (97 percent) patients reported nonmotor symptoms, with each patient experiencing an average of approximately eight nonmotor symptoms [34]. Nonmotor symptoms in the psychiatric domain occurred most frequently. Psychiatric symptoms such as psychosis or dementia may cause more disability than the motor features and may be more difficult to treat. In a single center survey of 265 patients with PD, pain, mood disorders, and sleep problems were the most troublesome nonmotor symptoms occurring in both early and late stage PD [37]. In some patients, certain nonmotor features of PD (eg, olfactory dysfunction, constipation, depression, and REM sleep behavior disorder) may present before the motor ones [5].")
27
DIAGNÓSTICO Diagnóstico clínico
Bradicinesia + 1 das manifestações cardinais. Início unilateral, assimetria persistente no curso da doença. “Gold standard” – exame neuropatológico. Exames complementares são de pouca relevância. DIAGNOSIS — The practical diagnosis of PD during life is based on clinical impression. There are no physiologic tests or blood tests for confirming the diagnosis, and neurodiagnostic testing with computerized imaging is almost always unrevealing. The true "gold standard" for diagnosis is neuropathologic examination. It is generally accepted that bradykinesia, plus one of the other two cardinal manifestations (tremor, rigidity) must be present in order to make the diagnosis of idiopathic PD. In addition, an excellent response to dopaminergic therapy is an important criterion for the diagnosis. Other clinical features that are supportive of the diagnosis are unilateral onset, presence of a rest tremor, and a persistent asymmetry throughout the course of the disease with the side of onset most affected
 must be present in order to make the diagnosis of idiopathic PD. In addition, an excellent response to dopaminergic therapy is an important criterion for the diagnosis. Other clinical features that are supportive of the diagnosis are unilateral onset, presence of a rest tremor, and a persistent asymmetry throughout the course of the disease with the side of onset most affected.")
28
DIAGNÓSTICO Resposta à terapia Dopaminérgica - Útil para diferenciar a Doença de Parkinson de outras Síndromes Parkinsonianas. - A Prova Terapêutica é considerada positiva quando há uma melhora no score UPDRS, de 15-30%, 1h após o uso de Levodopa. Response to dopaminergic therapy — As noted above, an excellent response to dopaminergic therapy is an important supportive feature for establishing the diagnosis of PD. The response to dopaminergic therapy in most parkinsonian syndromes is reduced or absent compared with the response in PD. However, up to 20 percent of patients with parkinsonism due to multiple system atrophy may respond initially to levodopa [33], as may a substantial proportion of those with vascular parkinsonism [34]. An acute dopaminergic challenge test consists of rater-blinded assessment of parkinsonian symptoms using the Unified Parkinson Disease Rating Scale (UPDRS) before and after a dose of levodopa (eg, carbidopa-levodopa 25/250 mg) or subcutaneous apomorphine (1.5 to 4.5 mg). Although there is no standard definition, a challenge is considered positive if there is a clinically significant improvement in the UPDRS score (usually in the range of 15 to 30 percent or more) one hour after levodopa administration or 20 minutes after apomorphine injection [35,36]. A systematic review and practice parameter from the American Academy of Neurology (AAN) published in 2006 concluded that levodopa and apomorphine challenge tests should be considered when the diagnosis of PD is uncertain, as both tests are "probably useful" in distinguishing PD from other parkinsonian syndromes
 before and after a dose of levodopa (eg, carbidopa-levodopa 25/250 mg) or subcutaneous apomorphine (1.5 to 4.5 mg). Although there is no standard definition, a challenge is considered positive if there is a clinically significant improvement in the UPDRS score (usually in the range of 15 to 30 percent or more) one hour after levodopa administration or 20 minutes after apomorphine injection [35,36]. A systematic review and practice parameter from the American Academy of Neurology (AAN) published in 2006 concluded that levodopa and apomorphine challenge tests should be considered when the diagnosis of PD is uncertain, as both tests are probably useful in distinguishing PD from other parkinsonian syndromes.")
29
DIAGNÓSTICO Características que sugerem diagnóstico alternativo: - Quedas ao início da doença - Ausência de resposta à Levodopa - Sinais motores simétricos - Demência que precede o quadro motor - Presença de Apraxia - Sinais cerebelares - Disautonomias no início do quadro Features suggesting an alternative diagnosis — According to the 2006 AAN systematic review and practice parameter, a number of clinical features in early stages of disease are probably useful for distinguishing other forms of parkinsonism from PD [37]: ●Falls at presentation or early in the course of the disease ●Poor response to levodopa ●Symmetrical motor signs ●Rapid progression to Hoehn and Yahr stage 3 (table 3) with mild to moderate disease and some postural instability, but physically independent ●Lack of tremor ●Dysautonomia, early in the disease course, as manifested by urinary urgency/incontinence and fecal incontinence, urinary retention requiring catheterization, persistent erectile failure, or symptomatic orthostatic hypotension Additional historical or clinical features that may suggest a diagnosis other than PD (table 1) include: ●History of encephalitis ●History of repeated head injury ●History of recurrent strokes and stepwise progression of parkinsonism ●Antipsychotic drug treatment at the onset of symptoms ●Presence of neoplasm or hydrocephalus on neuroimaging ●Cerebellar signs ●Supranuclear gaze palsy ●Dementia preceding or occurring concurrently with parkinsonism ●Babinski sign ●Presence of apraxia ●Strictly unilateral features after three years
 with mild to moderate disease and some postural instability, but physically independent. ●Lack of tremor. ●Dysautonomia, early in the disease course, as manifested by urinary urgency/incontinence and fecal incontinence, urinary retention requiring catheterization, persistent erectile failure, or symptomatic orthostatic hypotension. Additional historical or clinical features that may suggest a diagnosis other than PD (table 1) include: ●History of encephalitis. ●History of repeated head injury. ●History of recurrent strokes and stepwise progression of parkinsonism. ●Antipsychotic drug treatment at the onset of symptoms. ●Presence of neoplasm or hydrocephalus on neuroimaging. ●Cerebellar signs. ●Supranuclear gaze palsy. ●Dementia preceding or occurring concurrently with parkinsonism. ●Babinski sign. ●Presence of apraxia. ●Strictly unilateral features after three years.")
30
DIAGNÓSTICO DIFERENCIAL
Tremor Essencial Demência por Corpos de Lewys Degeneração Corticobasal Atrofia de Múltiplos Sistemas Parkinsonismo Induzido por Drogas Parkinsonismo Vascular DIFFERENTIAL DIAGNOSIS Essential tremor SWEDD Dementia with Lewy bodies Corticobasal degeneration Multiple system atrophy Progressive supranuclear palsy Idiopathic basal ganglia calcification Other neurodegenerative disorders Secondary parkinsonism Drug-induced parkinsonism Dementia with Lewy bodies — Dementia with Lewy bodies (DLB) is the second most common cause of neurodegenerative dementia after Alzheimer disease and is characterized clinically by visual hallucinations, fluctuating cognition, and parkinsonism. Other associated symptoms include repeated falls, syncope, autonomic dysfunction, neuroleptic sensitivity, delusions, hallucinations in nonvisual modalities, sleep disorders, and depression. (See "Clinical features and diagnosis of dementia with Lewy bodies".) Approximately 40 percent of patients with PD eventually develop dementia, and the differentiation of Parkinson disease dementia (PDD) from DLB is somewhat arbitrary. In PDD, dementia occurs in the setting of well established parkinsonism, while in DLB, dementia usually occurs concomitantly with or before the development of parkinsonian signs. Thus, patients are classified as having PDD if parkinsonism is present for more than one year before the onset of dementia. (See "Clinical features and diagnosis of dementia with Lewy bodies" and "Parkinson disease dementia".) Corticobasal degeneration — Patients with corticobasal degeneration can have asymmetric parkinsonism including bradykinesia, rigidity, and postural instability. More distinctive features can include ideomotor apraxia, alien limb phenomenon, aphasia, and loss of cortical sensory function [7]. Absence of tremor and lack of levodopa response are typical for corticobasal degeneration and help to distinguish it from PD. Multiple system atrophy commonly presents with parkinsonism, but patients also have varying degrees of dysautonomia, cerebellar involvement, and pyramidal signs. The prominence of these manifestations along with symmetry of onset, absence of tremor, and poor response to levodopa suggest this diagnosis rather than PD. However, some cases of multiple system atrophy may demonstrate responsiveness to levodopa, including motor fluctuations and dyskinesia, early in the course of the disease, with declining benefit over time. Cognitive function in multiple system atrophy tends to be relatively well preserved compared with PD and other parkinsonian syndromes, probably reflecting a lesser degree of cortical involvement
 is the second most common cause of neurodegenerative dementia after Alzheimer disease and is characterized clinically by visual hallucinations, fluctuating cognition, and parkinsonism. Other associated symptoms include repeated falls, syncope, autonomic dysfunction, neuroleptic sensitivity, delusions, hallucinations in nonvisual modalities, sleep disorders, and depression. (See Clinical features and diagnosis of dementia with Lewy bodies .) Approximately 40 percent of patients with PD eventually develop dementia, and the differentiation of Parkinson disease dementia (PDD) from DLB is somewhat arbitrary. In PDD, dementia occurs in the setting of well established parkinsonism, while in DLB, dementia usually occurs concomitantly with or before the development of parkinsonian signs. Thus, patients are classified as having PDD if parkinsonism is present for more than one year before the onset of dementia. (See Clinical features and diagnosis of dementia with Lewy bodies and Parkinson disease dementia .) Corticobasal degeneration — Patients with corticobasal degeneration can have asymmetric parkinsonism including bradykinesia, rigidity, and postural instability. More distinctive features can include ideomotor apraxia, alien limb phenomenon, aphasia, and loss of cortical sensory function [7]. Absence of tremor and lack of levodopa response are typical for corticobasal degeneration and help to distinguish it from PD. Multiple system atrophy commonly presents with parkinsonism, but patients also have varying degrees of dysautonomia, cerebellar involvement, and pyramidal signs. The prominence of these manifestations along with symmetry of onset, absence of tremor, and poor response to levodopa suggest this diagnosis rather than PD. However, some cases of multiple system atrophy may demonstrate responsiveness to levodopa, including motor fluctuations and dyskinesia, early in the course of the disease, with declining benefit over time. Cognitive function in multiple system atrophy tends to be relatively well preserved compared with PD and other parkinsonian syndromes, probably reflecting a lesser degree of cortical involvement.")
31
DIAGNÓSTICO DIFERENCIAL
Secondary parkinsonism — A wide variety of conditions can cause secondary parkinsonism, including the following [1]: ●Drugs (eg, classic and atypical antipsychotic agents, metoclopramide, prochlorperazine, reserpine). ●Toxins (eg, carbon disulfide, carbon monoxide, cyanide, MPTP, manganese, organic solvents). ●Head trauma, isolated or repeated (eg, boxing). ●Structural brain lesions that affect striatonigral circuits (eg, hydrocephalus, chronic subdural hematoma, tumor). (See "Normal pressure hydrocephalus" and "Clinical presentation and diagnosis of brain tumors".) ●Metabolic and miscellaneous disorders (eg, Wilson's disease, hypoparathyroidism and pseudohypoparathyroidism, chronic liver failure, extrapontine myelinolysis, neurodegeneration with brain iron accumulation, neuroacanthocytosis). (See "Wilson disease: Epidemiology and pathogenesis" and "Clinical manifestations of hypocalcemia" and "Bradykinetic movement disorders in children", section on 'Neurodegeneration with brain iron accumulation' and "Spiculated cells (echinocytes and acanthocytes) and target cells".) ●Infections (eg, encephalitis lethargica or Economo's encephalitis, HIV/AIDS, neurosyphilis, prion disease, progressive multifocal leukoencephalopathy, toxoplasmosis). (See "Approach to HIV-infected patients with central nervous system lesions" and "Neurosyphilis" and "Diseases of the central nervous system caused by prions" and "Progressive multifocal leukoencephalopathy: Epidemiology, clinical manifestations, and diagnosis".) ●Cerebrovascular disease. One theory holds that small vessel disease, particularly multiple lacunar infarcts in the basal ganglia and/or Binswanger disease, causes a "vascular parkinsonism". It should be noted that this entity is controversial [1], in part because most basal ganglia infarcts are not associated with parkinsonian signs [26,27]. Nevertheless, neuropathologic evidence suggests that mild parkinsonian signs in old age, particularly parkinsonian gait, are associated with the presence of macroscopic infarcts, microscopic infarcts, and arteriolosclerosis (ie, small vessel disease) [28]. (See "Lacunar infarcts".) Generally, the clinical history, associated features, and laboratory or radiologic findings in these cases allow the clinician to distinguish secondary parkinsonism and its underlying cause from PD or other primary parkinsonian syndromes. Drug-induced parkinsonism — Of the conditions causing secondary parkinsonism, drug-induced parkinsonism is the most common, and antipsychotic and antiemetics drugs are the most frequent offenders [1,29]. (See "First-generation antipsychotic medications: Pharmacology, administration, and comparative side effects" and "Second-generation antipsychotic medications: Pharmacology, administration, and comparative side effects".) Movement disorders such as akathisia and orofacial dyskinesia may be associated with chronic neuroleptic use and, if present, can be useful for distinguishing drug-induced parkinsonism from PD [30,31]. However, drug-induced parkinsonism can have clinical features identical to PD, including asymmetric onset with rest tremor [1]. (See "Tardive dyskinesia: Clinical features and diagnosis".) While drug-induced parkinsonism is usually reversible
. ●Toxins (eg, carbon disulfide, carbon monoxide, cyanide, MPTP, manganese, organic solvents). ●Head trauma, isolated or repeated (eg, boxing). ●Structural brain lesions that affect striatonigral circuits (eg, hydrocephalus, chronic subdural hematoma, tumor). (See Normal pressure hydrocephalus and Clinical presentation and diagnosis of brain tumors .) ●Metabolic and miscellaneous disorders (eg, Wilson s disease, hypoparathyroidism and pseudohypoparathyroidism, chronic liver failure, extrapontine myelinolysis, neurodegeneration with brain iron accumulation, neuroacanthocytosis). (See Wilson disease: Epidemiology and pathogenesis and Clinical manifestations of hypocalcemia and Bradykinetic movement disorders in children , section on Neurodegeneration with brain iron accumulation and Spiculated cells (echinocytes and acanthocytes) and target cells .) ●Infections (eg, encephalitis lethargica or Economo s encephalitis, HIV/AIDS, neurosyphilis, prion disease, progressive multifocal leukoencephalopathy, toxoplasmosis). (See Approach to HIV-infected patients with central nervous system lesions and Neurosyphilis and Diseases of the central nervous system caused by prions and Progressive multifocal leukoencephalopathy: Epidemiology, clinical manifestations, and diagnosis .) ●Cerebrovascular disease. One theory holds that small vessel disease, particularly multiple lacunar infarcts in the basal ganglia and/or Binswanger disease, causes a vascular parkinsonism . It should be noted that this entity is controversial [1], in part because most basal ganglia infarcts are not associated with parkinsonian signs [26,27]. Nevertheless, neuropathologic evidence suggests that mild parkinsonian signs in old age, particularly parkinsonian gait, are associated with the presence of macroscopic infarcts, microscopic infarcts, and arteriolosclerosis (ie, small vessel disease) [28]. (See Lacunar infarcts .) Generally, the clinical history, associated features, and laboratory or radiologic findings in these cases allow the clinician to distinguish secondary parkinsonism and its underlying cause from PD or other primary parkinsonian syndromes. Drug-induced parkinsonism — Of the conditions causing secondary parkinsonism, drug-induced parkinsonism is the most common, and antipsychotic and antiemetics drugs are the most frequent offenders [1,29]. (See First-generation antipsychotic medications: Pharmacology, administration, and comparative side effects and Second-generation antipsychotic medications: Pharmacology, administration, and comparative side effects .) Movement disorders such as akathisia and orofacial dyskinesia may be associated with chronic neuroleptic use and, if present, can be useful for distinguishing drug-induced parkinsonism from PD [30,31]. However, drug-induced parkinsonism can have clinical features identical to PD, including asymmetric onset with rest tremor [1]. (See Tardive dyskinesia: Clinical features and diagnosis .) While drug-induced parkinsonism is usually reversible.")
32
EXAMES COMPLEMENTARES
Ressonância Magnética PETscan DaTscan Ultrassonografia Transcraniana Teste olfatório ório

33
TRATAMENTO Medidas Gerais: - Educação - Suporte - Fisioterapia e Exercícios - Terapia da Fala - Terapia Nutricional INTRODUCTION — Parkinson disease (PD) is a chronic disorder that requires broad-based management including patient and family education, support group services, general wellness maintenance, exercise, and nutrition. EDUCATION — The prospect of having a chronic and progressive neurologic disease is frightening. Many individuals are familiar with Parkinson disease (PD) and may even have had first-hand acquaintance with its disabling effects in an affected family member or friend. Education is essential in order to provide the patient and family with some understanding and control over the disorder. SUPPORT — The emotional and psychologic needs of the patient with PD and family should be addressed. Normal reactions of anger, depression, anxiety, and social and economic concerns often begin with the onset of the disease and evolve as it progresses. . EXERCISE AND PHYSICAL THERAPY — Regular exercise promotes a feeling of physical and mental well-being; it is especially valuable due to the chronic nature of PD and its associated progressive motor limitations. Exercise may not slow the progression of akinesia, rigidity, or gait disturbance, but it can alleviate some secondary orthopedic effects of rigidity and flexed posture such as shoulder, hip, and back pain, and it may also improve function in some motor tasks [5-7]. Mounting evidence suggests that regular aerobic exercise has a positive impact on PD [7-10]. A 2013 systematic review observed that most of the benefits with physical therapy for patients with PD were small [19]. Nevertheless, many patients gain lasting confidence and a sense of control over one aspect of the disease, especially if they have never engaged in physical activity in the past. Referral to a physical therapist or exercise group is a good way to get patients started in such activities. SPEECH THERAPY — Dysarthria and hypophonia are common manifestations of PD. The practice parameter from the American Academy of Neurology (AAN) issued in 2006 concluded that speech therapy for patients with PD may be helpful in improving speech volume [6]. NUTRITION — Elderly patients with chronic illness are at risk for poor nutrition and weight loss. Prompt recognition and management of this problem is important to avoid loss of bone and muscle mass. No specific diet influences the course of PD, although certain recommendations can be made [23]. ●A high fiber diet and adequate hydration help manage the constipation of PD. ●Large, high-fat meals that slow gastric emptying and interfere with medication absorption should be avoided. ●Dietary protein restriction is not necessary except in some patients with advanced disease and motor fluctuations in whom competition with other amino acids interferes with L-dopa absorption.
 is a chronic disorder that requires broad-based management including patient and family education, support group services, general wellness maintenance, exercise, and nutrition. EDUCATION — The prospect of having a chronic and progressive neurologic disease is frightening. Many individuals are familiar with Parkinson disease (PD) and may even have had first-hand acquaintance with its disabling effects in an affected family member or friend. Education is essential in order to provide the patient and family with some understanding and control over the disorder. SUPPORT — The emotional and psychologic needs of the patient with PD and family should be addressed. Normal reactions of anger, depression, anxiety, and social and economic concerns often begin with the onset of the disease and evolve as it progresses. . EXERCISE AND PHYSICAL THERAPY — Regular exercise promotes a feeling of physical and mental well-being; it is especially valuable due to the chronic nature of PD and its associated progressive motor limitations. Exercise may not slow the progression of akinesia, rigidity, or gait disturbance, but it can alleviate some secondary orthopedic effects of rigidity and flexed posture such as shoulder, hip, and back pain, and it may also improve function in some motor tasks [5-7]. Mounting evidence suggests that regular aerobic exercise has a positive impact on PD [7-10]. A 2013 systematic review observed that most of the benefits with physical therapy for patients with PD were small [19]. Nevertheless, many patients gain lasting confidence and a sense of control over one aspect of the disease, especially if they have never engaged in physical activity in the past. Referral to a physical therapist or exercise group is a good way to get patients started in such activities. SPEECH THERAPY — Dysarthria and hypophonia are common manifestations of PD. The practice parameter from the American Academy of Neurology (AAN) issued in 2006 concluded that speech therapy for patients with PD may be helpful in improving speech volume [6]. NUTRITION — Elderly patients with chronic illness are at risk for poor nutrition and weight loss. Prompt recognition and management of this problem is important to avoid loss of bone and muscle mass. No specific diet influences the course of PD, although certain recommendations can be made [23]. ●A high fiber diet and adequate hydration help manage the constipation of PD. ●Large, high-fat meals that slow gastric emptying and interfere with medication absorption should be avoided. ●Dietary protein restriction is not necessary except in some patients with advanced disease and motor fluctuations in whom competition with other amino acids interferes with L-dopa absorption.")
34
TRATAMENTO FARMACOLÓGICO
Levodopa Agonistas da Dopamina – bromocriptina, apomorfina Inibidores da MAO – selegilina, rasgilina Anticolinérgicos – triexifenidil, benztropina Amantadina Inibidores da COMT – tolcapone, entacapone The major drugs available for the treatment of PD motor symptoms include: ●Levodopa ●Dopamine agonists ●MAO B inhibitors ●Anticholinergic agents ●Amantadine ●COMT inhibitors The following general principles can be used to guide the choice of therapy in symptomatic PD: ●Levodopa is the most effective drug for the symptomatic treatment of PD and is the drug of first choice if symptoms ●The dopamine agonists may be employed either as monotherapy in early PD or in combination with other antiparkinsonian drugs for treatment of more advanced disease. ●Either levodopa or a dopamine agonist can be used initially for patients who require symptomatic therapy for PD [2,3 ●The MAO B inhibitors selegiline and rasagiline may be useful in patients with early PD but have only modest symptomatic benefit as monotherapy. (See 'MAO B inhibitors' below.) ●Anticholinergic drugs are most useful as monotherapy in patients under 70 years of age with disturbing tremor who do not have significant bradykinesia or gait disturbance. They also may be useful in patients with more advanced disease who have persistent tremor despite treatment with levodopa or dopamine agonists. Their use in older or demented individuals and those without tremor is strongly discouraged. (See 'Anticholinergics' below.) ●Amantadine is a relatively weak antiparkinsonian drug with low toxicity that is most useful in treating younger patients with early or mild PD and perhaps later when dyskinesia becomes problematic. ●Low-dose estrogen may be helpful as adjunctive therapy in postmenopausal women Parkinsonism-hyperpyrexia syndrome — There have been reports of patients with PD who developed neuroleptic malignant syndrome in the context of sudden withdrawal or dose reductions of levodopa or dopamine agonists
 ●Anticholinergic drugs are most useful as monotherapy in patients under 70 years of age with disturbing tremor who do not have significant bradykinesia or gait disturbance. They also may be useful in patients with more advanced disease who have persistent tremor despite treatment with levodopa or dopamine agonists. Their use in older or demented individuals and those without tremor is strongly discouraged. (See Anticholinergics below.) ●Amantadine is a relatively weak antiparkinsonian drug with low toxicity that is most useful in treating younger patients with early or mild PD and perhaps later when dyskinesia becomes problematic. ●Low-dose estrogen may be helpful as adjunctive therapy in postmenopausal women. Parkinsonism-hyperpyrexia syndrome — There have been reports of patients with PD who developed neuroleptic malignant syndrome in the context of sudden withdrawal or dose reductions of levodopa or dopamine agonists.")
35
TRATAMENTO FARMACOLÓGICO
LEVODOPA Iniciar o tratamento com meio comprimido de cardiodopa-levodopa 25/100mg, 3x ao dia. Aumentar progressivamente a dose, até que o consumo diário fique entre mg/dia. LEVODOPA — Levodopa (L-dopa) is well-established as the most effective drug for the symptomatic treatment of idiopathic or Lewy body PD [2,7]. It is particularly effective for the management of bradykinetic symptoms and should be introduced when these become intrusive or troublesome or are uncontrolled by other antiparkinsonian drugs. Formulations — Levodopa is combined with a peripheral decarboxylase inhibitor to block its conversion to dopamine in the systemic circulation and liver (before it crosses the blood-brain barrier) in order to prevent nausea, vomiting, and orthostatic hypotension. In the United States, the decarboxylase inhibitor is carbidopa. The combination drug carbidopa-levodopa (immediate-release Sinemet) is available in tablets of 10/100, 25/100, and 25/250 mg, with the numerator referring to carbidopa and the denominator referring to the levodopa dose. Dosing — Treatment should begin with small doses, such as carbidopa-levodopa (Sinemet) 25/100 mg, one-half tablet two to three times daily with meals. The usual practice is to titrate to the lowest levodopa dose that produces a useful clinical response. This varies from patient to patient, but at the start it is typically in the vicinity of 300 to 600 mg of levodopa daily. The vast majority of patients with idiopathic PD will enjoy a significant therapeutic response to moderate doses of levodopa (300 to 600 mg daily). Complete absence of response to a levodopa dose of 1000 to 1500 mg/day suggests that the original diagnosis of PD may be incorrect and that one of the other parkinsonian syndromes, such as multiple system atrophy, progressive supranuclear palsy, or vascular parkinsonism should be considered. (See "Diagnosis of Parkinson disease".) Adverse effects — Nausea, somnolence, dizziness, and headache are among the more common early side effects. More serious adverse reactions to levodopa may include confusion, hallucinations, delusions, agitation, psychosis, and orthostatic hypotension. Motor fluctuations — involuntary movements known as dyskinesia, abnormal cramps and postures of the extremities and trunk known as dystonia, and a variety of complex fluctuations in motor function [18,19]. It is estimated that such motor complications occur in at least 50 percent of patients after 5 to 10 years of treatment [1 The choice of initial therapy for PD – whether levodopa, dopamine agonist, or MAO B inhibitor – has little impact on the long-term outcome of PD in terms of motor fluctuations and dyskinesia [28-30]. T he increase in motor fluctuations over time is most likely due to progressive degeneration of nigrostriatal dopamine terminals, which increasingly limits the normal physiologic uptake and release of dopamine, thereby leading to reduced buffering of the natural fluctuations in plasma levodopa levels that occur due to its 90-minute pharmacologic half-life [1 There has been longstanding concern among some clinicians that levodopa causes motor fluctuations and dyskinesia by its potential to promote oxidative stress and accelerated neurodegeneration, rather than by the change in levodopa pharmacodynamics that occurs with natural progression of the underlying disease [31,32]. Therefore, it is commonly proposed that the initiation of levodopa be delayed until symptoms significantly interfere with function. Others contend, however, that there is no convincing evidence that levodopa is itself responsible for late motor complications, and that delay of treatment unnecessarily deprives patients of therapeutic benefit early in the disease, when the potential for sustained improvement is greatest [33]. Neurotoxic versus neuroprotective effects — The concern that prolonged use of levodopa may directly hasten the degeneration of dopamine neurons in the substantia nigra by promoting the generation of free radicals and oxidative stress is one rationale for delaying the use of levodopa in the treatment of PD [34]. Another more compelling reason for delaying the use of levodopa is to possibly postpone the appearance of motor complications such as dyskinesia and motor fluctuations. The evidence regarding potential effects of levodopa on dopaminergic neurons is summarized by the following observations: In conclusion, the debate over the potential neuroprotective versus neurotoxic effects of levodopa remains unresolved [43,44], but the weight of the evidence accumulated to date shows that long term use of levodopa is not a hazard, especially in light of its superiority over all other pharmacotherapies.
 is well-established as the most effective drug for the symptomatic treatment of idiopathic or Lewy body PD [2,7]. It is particularly effective for the management of bradykinetic symptoms and should be introduced when these become intrusive or troublesome or are uncontrolled by other antiparkinsonian drugs. Formulations — Levodopa is combined with a peripheral decarboxylase inhibitor to block its conversion to dopamine in the systemic circulation and liver (before it crosses the blood-brain barrier) in order to prevent nausea, vomiting, and orthostatic hypotension. In the United States, the decarboxylase inhibitor is carbidopa. The combination drug carbidopa-levodopa (immediate-release Sinemet) is available in tablets of 10/100, 25/100, and 25/250 mg, with the numerator referring to carbidopa and the denominator referring to the levodopa dose. Dosing — Treatment should begin with small doses, such as carbidopa-levodopa (Sinemet) 25/100 mg, one-half tablet two to three times daily with meals. The usual practice is to titrate to the lowest levodopa dose that produces a useful clinical response. This varies from patient to patient, but at the start it is typically in the vicinity of 300 to 600 mg of levodopa daily. The vast majority of patients with idiopathic PD will enjoy a significant therapeutic response to moderate doses of levodopa (300 to 600 mg daily). Complete absence of response to a levodopa dose of 1000 to 1500 mg/day suggests that the original diagnosis of PD may be incorrect and that one of the other parkinsonian syndromes, such as multiple system atrophy, progressive supranuclear palsy, or vascular parkinsonism should be considered. (See Diagnosis of Parkinson disease .) Adverse effects — Nausea, somnolence, dizziness, and headache are among the more common early side effects. More serious adverse reactions to levodopa may include confusion, hallucinations, delusions, agitation, psychosis, and orthostatic hypotension. Motor fluctuations — involuntary movements known as dyskinesia, abnormal cramps and postures of the extremities and trunk known as dystonia, and a variety of complex fluctuations in motor function [18,19]. It is estimated that such motor complications occur in at least 50 percent of patients after 5 to 10 years of treatment [1. The choice of initial therapy for PD – whether levodopa, dopamine agonist, or MAO B inhibitor – has little impact on the long-term outcome of PD in terms of motor fluctuations and dyskinesia [28-30]. T. he increase in motor fluctuations over time is most likely due to progressive degeneration of nigrostriatal dopamine terminals, which increasingly limits the normal physiologic uptake and release of dopamine, thereby leading to reduced buffering of the natural fluctuations in plasma levodopa levels that occur due to its 90-minute pharmacologic half-life [1. There has been longstanding concern among some clinicians that levodopa causes motor fluctuations and dyskinesia by its potential to promote oxidative stress and accelerated neurodegeneration, rather than by the change in levodopa pharmacodynamics that occurs with natural progression of the underlying disease [31,32]. Therefore, it is commonly proposed that the initiation of levodopa be delayed until symptoms significantly interfere with function. Others contend, however, that there is no convincing evidence that levodopa is itself responsible for late motor complications, and that delay of treatment unnecessarily deprives patients of therapeutic benefit early in the disease, when the potential for sustained improvement is greatest [33]. Neurotoxic versus neuroprotective effects — The concern that prolonged use of levodopa may directly hasten the degeneration of dopamine neurons in the substantia nigra by promoting the generation of free radicals and oxidative stress is one rationale for delaying the use of levodopa in the treatment of PD [34]. Another more compelling reason for delaying the use of levodopa is to possibly postpone the appearance of motor complications such as dyskinesia and motor fluctuations. The evidence regarding potential effects of levodopa on dopaminergic neurons is summarized by the following observations: In conclusion, the debate over the potential neuroprotective versus neurotoxic effects of levodopa remains unresolved [43,44], but the weight of the evidence accumulated to date shows that long term use of levodopa is not a hazard, especially in light of its superiority over all other pharmacotherapies.")
36
Deep Brain Stimulation – STN ou GPi
TRATAMENTO CIRURGÍCO Deep Brain Stimulation – STN ou GPi Talamotomia – Núcleo Ventral intermédio Palidotomia – porção póstero-lateral Subtalamotomia Infusão de GDNF Transplante Neural Terapia Gênica Infusão Duodenal de Levodopa Rationale for surgery — Two downstream effects of nigral degeneration and dopamine deficiency in PD are excessive subthalamic nucleus (STN) excitation of the internal globus pallidus (GPi) and excessive globus pallidus inhibition of the thalamus. These, in turn, cause reduced thalamocortical activity, which is believed to mediate the symptoms of akinesia and rigidity. Pallidotomy reverses the excess pallidal inhibitory effect and improves parkinsonism by targeted ablation of the GPi. High frequency deep brain stimulation (DBS) suppresses neuronal activity and also activates efferent fiber pathways leaving the targeted nucleus. Thalamotomy and pallidotomy are immediate in effect but irreversible; DBS makes no major lesion, but it requires intensive adjustments in the postoperative period and lifelong maintenance with risk of hardware complications and infection. Since DBS produces a safe and entirely reversible physiologic effect without destroying brain tissue, DBS is now being used in place of lesions (pallidotomy and thalamotomy) to suppress excessive GPi or STN activity. DEEP BRAIN STIMULATION — Deep brain stimulation (DBS) is currently the most frequently performed surgical procedure for the treatment of advanced PD [2]. The use of DBS for patients with shorter duration PD is also being explored. Deep brain stimulation (DBS) of the subthalamic nucleus (STN) and globus pallidus pars interna (GPi) are both effective for improving PD motor symptoms and appear to have similar safety. However, STN DBS leads to greater reduction in antiparkinson medication doses. Evidence from randomized controlled trials supports the conclusion that DBS of either the STN or the GPi alleviates the motor fluctuations and dyskinesia associated with advanced PD. Both STN and GPi stimulation are equally effective for motor symptoms including tremor and bradykinesia [5]. Antiparkinson medication reduction is greater after STN stimulation, while GPi stimulation may be superior for control of dyskinesia. Although the evidence is inconsistent, some studies suggest GPi stimulation is associated with fewer complications related to cognitive, mood, and behavioral problems [5,45]. The long-term durability of both procedures needs to be further investigated.
 excitation of the internal globus pallidus (GPi) and excessive globus pallidus inhibition of the thalamus. These, in turn, cause reduced thalamocortical activity, which is believed to mediate the symptoms of akinesia and rigidity. Pallidotomy reverses the excess pallidal inhibitory effect and improves parkinsonism by targeted ablation of the GPi. High frequency deep brain stimulation (DBS) suppresses neuronal activity and also activates efferent fiber pathways leaving the targeted nucleus. Thalamotomy and pallidotomy are immediate in effect but irreversible; DBS makes no major lesion, but it requires intensive adjustments in the postoperative period and lifelong maintenance with risk of hardware complications and infection. Since DBS produces a safe and entirely reversible physiologic effect without destroying brain tissue, DBS is now being used in place of lesions (pallidotomy and thalamotomy) to suppress excessive GPi or STN activity. DEEP BRAIN STIMULATION — Deep brain stimulation (DBS) is currently the most frequently performed surgical procedure for the treatment of advanced PD [2]. The use of DBS for patients with shorter duration PD is also being explored. Deep brain stimulation (DBS) of the subthalamic nucleus (STN) and globus pallidus pars interna (GPi) are both effective for improving PD motor symptoms and appear to have similar safety. However, STN DBS leads to greater reduction in antiparkinson medication doses. Evidence from randomized controlled trials supports the conclusion that DBS of either the STN or the GPi alleviates the motor fluctuations and dyskinesia associated with advanced PD. Both STN and GPi stimulation are equally effective for motor symptoms including tremor and bradykinesia [5]. Antiparkinson medication reduction is greater after STN stimulation, while GPi stimulation may be superior for control of dyskinesia. Although the evidence is inconsistent, some studies suggest GPi stimulation is associated with fewer complications related to cognitive, mood, and behavioral problems [5,45]. The long-term durability of both procedures needs to be further investigated.")
37
Deep Brain Stimulation – STN ou GPi
TRATAMENTO CIRURGICO Deep Brain Stimulation – STN ou GPi Talamotomia – Núcleo Ventral intermédio Palidotomia – porção póstero-lateral Rationale for surgery — Two downstream effects of nigral degeneration and dopamine deficiency in PD are excessive subthalamic nucleus (STN) excitation of the internal globus pallidus (GPi) and excessive globus pallidus inhibition of the thalamus. These, in turn, cause reduced thalamocortical activity, which is believed to mediate the symptoms of akinesia and rigidity. Pallidotomy reverses the excess pallidal inhibitory effect and improves parkinsonism by targeted ablation of the GPi. High frequency deep brain stimulation (DBS) suppresses neuronal activity and also activates efferent fiber pathways leaving the targeted nucleus. Thalamotomy and pallidotomy are immediate in effect but irreversible; DBS makes no major lesion, but it requires intensive adjustments in the postoperative period and lifelong maintenance with risk of hardware complications and infection. Since DBS produces a safe and entirely reversible physiologic effect without destroying brain tissue, DBS is now being used in place of lesions (pallidotomy and thalamotomy) to suppress excessive GPi or STN activity. DEEP BRAIN STIMULATION — Deep brain stimulation (DBS) is currently the most frequently performed surgical procedure for the treatment of advanced PD [2]. The use of DBS for patients with shorter duration PD is also being explored. Deep brain stimulation (DBS) of the subthalamic nucleus (STN) and globus pallidus pars interna (GPi) are both effective for improving PD motor symptoms and appear to have similar safety. However, STN DBS leads to greater reduction in antiparkinson medication doses. Evidence from randomized controlled trials supports the conclusion that DBS of either the STN or the GPi alleviates the motor fluctuations and dyskinesia associated with advanced PD. Both STN and GPi stimulation are equally effective for motor symptoms including tremor and bradykinesia [5]. Antiparkinson medication reduction is greater after STN stimulation, while GPi stimulation may be superior for control of dyskinesia. Although the evidence is inconsistent, some studies suggest GPi stimulation is associated with fewer complications related to cognitive, mood, and behavioral problems [5,45]. The long-term durability of both procedures needs to be further investigated. Unilateral pallidotomy is effective and relatively safe compared with medical therapy in patients with severe dyskinesia and "on" and "off" fluctuations [55,56]. Unilateral thalamotomy and DBS of the ventral intermediate nucleus of the thalamus can safely and effectively control tremor in selected cases of PD. However, they do not help bradykinesia, and since this typically becomes the most important symptom over time, GPi and STN should be considered the targets of choice in patients with PD even if tremor is the main symptom
 excitation of the internal globus pallidus (GPi) and excessive globus pallidus inhibition of the thalamus. These, in turn, cause reduced thalamocortical activity, which is believed to mediate the symptoms of akinesia and rigidity. Pallidotomy reverses the excess pallidal inhibitory effect and improves parkinsonism by targeted ablation of the GPi. High frequency deep brain stimulation (DBS) suppresses neuronal activity and also activates efferent fiber pathways leaving the targeted nucleus. Thalamotomy and pallidotomy are immediate in effect but irreversible; DBS makes no major lesion, but it requires intensive adjustments in the postoperative period and lifelong maintenance with risk of hardware complications and infection. Since DBS produces a safe and entirely reversible physiologic effect without destroying brain tissue, DBS is now being used in place of lesions (pallidotomy and thalamotomy) to suppress excessive GPi or STN activity. DEEP BRAIN STIMULATION — Deep brain stimulation (DBS) is currently the most frequently performed surgical procedure for the treatment of advanced PD [2]. The use of DBS for patients with shorter duration PD is also being explored. Deep brain stimulation (DBS) of the subthalamic nucleus (STN) and globus pallidus pars interna (GPi) are both effective for improving PD motor symptoms and appear to have similar safety. However, STN DBS leads to greater reduction in antiparkinson medication doses. Evidence from randomized controlled trials supports the conclusion that DBS of either the STN or the GPi alleviates the motor fluctuations and dyskinesia associated with advanced PD. Both STN and GPi stimulation are equally effective for motor symptoms including tremor and bradykinesia [5]. Antiparkinson medication reduction is greater after STN stimulation, while GPi stimulation may be superior for control of dyskinesia. Although the evidence is inconsistent, some studies suggest GPi stimulation is associated with fewer complications related to cognitive, mood, and behavioral problems [5,45]. The long-term durability of both procedures needs to be further investigated. Unilateral pallidotomy is effective and relatively safe compared with medical therapy in patients with severe dyskinesia and on and off fluctuations [55,56]. Unilateral thalamotomy and DBS of the ventral intermediate nucleus of the thalamus can safely and effectively control tremor in selected cases of PD. However, they do not help bradykinesia, and since this typically becomes the most important symptom over time, GPi and STN should be considered the targets of choice in patients with PD even if tremor is the main symptom.")
38
TRATAMENTO CIRURGICO This schematic drawing of the basal ganglia and their connections illustrates the main striato-fugal pathways. In Parkinson disease (PD), the indirect inhibitory pathway mediated via D2 striatal receptors is postulated to be overactive, whereas the direct inhibitory pathway mediated via D1 striatal receptors is underactive. As a result, the main pallidal-thalamic outflow pathway provides excessive inhibitory input to the thalamus, which causes suppression of thalamo-cortical-spinal pathway, manifested clinically by paucity and slowness of movement (bradykinesia). Green: excitatory; Red: inhibitory; GPe: globus pallidus externa; GPi: globus pallidus interna; SNc: substantia nigra pars compacta; SNr: substantia nigra pars reticulata; STN: subthalamic nucleus; TH: ventrolateral nucleus of the thalamus. Because of the apparent discrepancy between loss of striatal dopamine (>80 percent) and the degree of loss of neurons in the substantia nigra (50 to 60 percent), some have suggested that the initial site of pathology is in the striatum and that retrograde degeneration may be responsible for the neuronal loss in the substantia nigra [3]. An alternative explanation is that each dopaminergic neuron has multiple projections that terminate in the striatum, so that death of the cell body has a multiplying effect on loss of terminals. Five distinct dopamine receptors (D1 through D5) have been cloned and characterized; they are found throughout the basal ganglia and limbic system. The D1 and D2 receptors are highly concentrated in the dorsal (motor) striatum and are the most relevant to the pathophysiology of PD because they are activated by the dopaminergic pathway originating in the SNc and terminating in the caudate and putamen. Receptors designated as D3, D4, and D5 are more abundant in the mesolimbic or emotional part of the brain (D3, D4) and hippocampus/hypothalamus (D5) [5]. ●The indirect pathway is mediated chiefly via dopamine's inhibitory influence on striatal D2 dopamine receptors. In the indirect pathway, the striatum projects to the neurons in the lateral GP (GPe) utilizing GABA, and the GPe in turn projects to the STN, which provides excitatory input via glutamate to the internal segment of the GP (GPi) and SN pars reticulata (SNr). GPi neurons are GABAergic and synapse in the ventrolateral nucleus of the thalamus. Thalamic input to the cortex is excitatory.
, the indirect inhibitory pathway mediated via D2 striatal receptors is postulated to be overactive, whereas the direct inhibitory pathway mediated via D1 striatal receptors is underactive. As a result, the main pallidal-thalamic outflow pathway provides excessive inhibitory input to the thalamus, which causes suppression of thalamo-cortical-spinal pathway, manifested clinically by paucity and slowness of movement (bradykinesia). Green: excitatory; Red: inhibitory; GPe: globus pallidus externa; GPi: globus pallidus interna; SNc: substantia nigra pars compacta; SNr: substantia nigra pars reticulata; STN: subthalamic nucleus; TH: ventrolateral nucleus of the thalamus. Because of the apparent discrepancy between loss of striatal dopamine (>80 percent) and the degree of loss of neurons in the substantia nigra (50 to 60 percent), some have suggested that the initial site of pathology is in the striatum and that retrograde degeneration may be responsible for the neuronal loss in the substantia nigra [3]. An alternative explanation is that each dopaminergic neuron has multiple projections that terminate in the striatum, so that death of the cell body has a multiplying effect on loss of terminals. Five distinct dopamine receptors (D1 through D5) have been cloned and characterized; they are found throughout the basal ganglia and limbic system. The D1 and D2 receptors are highly concentrated in the dorsal (motor) striatum and are the most relevant to the pathophysiology of PD because they are activated by the dopaminergic pathway originating in the SNc and terminating in the caudate and putamen. Receptors designated as D3, D4, and D5 are more abundant in the mesolimbic or emotional part of the brain (D3, D4) and hippocampus/hypothalamus (D5) [5]. ●The indirect pathway is mediated chiefly via dopamine s inhibitory influence on striatal D2 dopamine receptors. In the indirect pathway, the striatum projects to the neurons in the lateral GP (GPe) utilizing GABA, and the GPe in turn projects to the STN, which provides excitatory input via glutamate to the internal segment of the GP (GPi) and SN pars reticulata (SNr). GPi neurons are GABAergic and synapse in the ventrolateral nucleus of the thalamus. Thalamic input to the cortex is excitatory.")
39
TRATAMENTO CIRURGICO This schematic drawing of the basal ganglia and their connections illustrates the main striato-fugal pathways. In Parkinson disease (PD), the indirect inhibitory pathway mediated via D2 striatal receptors is postulated to be overactive, whereas the direct inhibitory pathway mediated via D1 striatal receptors is underactive. As a result, the main pallidal-thalamic outflow pathway provides excessive inhibitory input to the thalamus, which causes suppression of thalamo-cortical-spinal pathway, manifested clinically by paucity and slowness of movement (bradykinesia). Green: excitatory; Red: inhibitory; GPe: globus pallidus externa; GPi: globus pallidus interna; SNc: substantia nigra pars compacta; SNr: substantia nigra pars reticulata; STN: subthalamic nucleus; TH: ventrolateral nucleus of the thalamus. Because of the apparent discrepancy between loss of striatal dopamine (>80 percent) and the degree of loss of neurons in the substantia nigra (50 to 60 percent), some have suggested that the initial site of pathology is in the striatum and that retrograde degeneration may be responsible for the neuronal loss in the substantia nigra [3]. An alternative explanation is that each dopaminergic neuron has multiple projections that terminate in the striatum, so that death of the cell body has a multiplying effect on loss of terminals. Five distinct dopamine receptors (D1 through D5) have been cloned and characterized; they are found throughout the basal ganglia and limbic system. The D1 and D2 receptors are highly concentrated in the dorsal (motor) striatum and are the most relevant to the pathophysiology of PD because they are activated by the dopaminergic pathway originating in the SNc and terminating in the caudate and putamen. Receptors designated as D3, D4, and D5 are more abundant in the mesolimbic or emotional part of the brain (D3, D4) and hippocampus/hypothalamus (D5) [5]. ●The indirect pathway is mediated chiefly via dopamine's inhibitory influence on striatal D2 dopamine receptors. In the indirect pathway, the striatum projects to the neurons in the lateral GP (GPe) utilizing GABA, and the GPe in turn projects to the STN, which provides excitatory input via glutamate to the internal segment of the GP (GPi) and SN pars reticulata (SNr). GPi neurons are GABAergic and synapse in the ventrolateral nucleus of the thalamus. Thalamic input to the cortex is excitatory.
, the indirect inhibitory pathway mediated via D2 striatal receptors is postulated to be overactive, whereas the direct inhibitory pathway mediated via D1 striatal receptors is underactive. As a result, the main pallidal-thalamic outflow pathway provides excessive inhibitory input to the thalamus, which causes suppression of thalamo-cortical-spinal pathway, manifested clinically by paucity and slowness of movement (bradykinesia). Green: excitatory; Red: inhibitory; GPe: globus pallidus externa; GPi: globus pallidus interna; SNc: substantia nigra pars compacta; SNr: substantia nigra pars reticulata; STN: subthalamic nucleus; TH: ventrolateral nucleus of the thalamus. Because of the apparent discrepancy between loss of striatal dopamine (>80 percent) and the degree of loss of neurons in the substantia nigra (50 to 60 percent), some have suggested that the initial site of pathology is in the striatum and that retrograde degeneration may be responsible for the neuronal loss in the substantia nigra [3]. An alternative explanation is that each dopaminergic neuron has multiple projections that terminate in the striatum, so that death of the cell body has a multiplying effect on loss of terminals. Five distinct dopamine receptors (D1 through D5) have been cloned and characterized; they are found throughout the basal ganglia and limbic system. The D1 and D2 receptors are highly concentrated in the dorsal (motor) striatum and are the most relevant to the pathophysiology of PD because they are activated by the dopaminergic pathway originating in the SNc and terminating in the caudate and putamen. Receptors designated as D3, D4, and D5 are more abundant in the mesolimbic or emotional part of the brain (D3, D4) and hippocampus/hypothalamus (D5) [5]. ●The indirect pathway is mediated chiefly via dopamine s inhibitory influence on striatal D2 dopamine receptors. In the indirect pathway, the striatum projects to the neurons in the lateral GP (GPe) utilizing GABA, and the GPe in turn projects to the STN, which provides excitatory input via glutamate to the internal segment of the GP (GPi) and SN pars reticulata (SNr). GPi neurons are GABAergic and synapse in the ventrolateral nucleus of the thalamus. Thalamic input to the cortex is excitatory.")
40
ALGORITMO

41
PROGNÓSTICO Progressão variada
Em 5 anos, 25% dos pacientes ficam severamente incapacitados ou vão a óbito. Em 10 anos, esse número chega à 77%. A prevalência de demência chega a 40% em 10 anos. A gravidade e variabilidade dos sintomas aumenta com a evolução da doença. DISEASE PROGRESSION AND PROGNOSIS — As noted above, progression in PD is variable, and there currently are no symptoms or signs in idiopathic PD that allow a practitioner to accurately predict the future course of PD for any given individual. (See 'Clinical subtypes' above.) However, several observations provide insight into the general rate of disease progression in PD. ●In an early landmark study of patients with PD seen from 1949 to 1964, the proportion who were severely disabled or dead within five years of disease onset was approximately 25 percent [16]. This increased to 67 percent at 5 to 9 years, and 80 percent at 10 to 14 years. A small group of atypical patients had slow disease progression, maintaining balance and postural stability for ≥10 years, and lacking severe disability even at ≥20 years. Results were similar in a later study of 142 patients with PD followed from 2000 to 2012, with approximately 77 percent having a poor outcome (ie, death, dementia, or postural instability) at 10 years after diagnosis [31]. ●A report of 618 patients with PD found that the transition from disease impairment to disability occurred generally between three and seven years after the onset of PD [134]. Impairment was defined by difficulty with daily activities without loss of independent function, while disability was defined by loss of independent function. ●Most studies suggest that mortality is only modestly increased for patients with PD compared with age-matched controls [31, ]. ●The prevalence of dementia in PD is approximately 40 percent, and increases with age and disease duration
 However, several observations provide insight into the general rate of disease progression in PD. ●In an early landmark study of patients with PD seen from 1949 to 1964, the proportion who were severely disabled or dead within five years of disease onset was approximately 25 percent [16]. This increased to 67 percent at 5 to 9 years, and 80 percent at 10 to 14 years. A small group of atypical patients had slow disease progression, maintaining balance and postural stability for ≥10 years, and lacking severe disability even at ≥20 years. Results were similar in a later study of 142 patients with PD followed from 2000 to 2012, with approximately 77 percent having a poor outcome (ie, death, dementia, or postural instability) at 10 years after diagnosis [31]. ●A report of 618 patients with PD found that the transition from disease impairment to disability occurred generally between three and seven years after the onset of PD [134]. Impairment was defined by difficulty with daily activities without loss of independent function, while disability was defined by loss of independent function. ●Most studies suggest that mortality is only modestly increased for patients with PD compared with age-matched controls [31, ]. ●The prevalence of dementia in PD is approximately 40 percent, and increases with age and disease duration.")
42
REFERÊNCIAS 1.Parkinson J. An Essay on the Shaking Palsy, Sherwood, Neely, and Jones, London Marras, C, Tanner, CM. Epidemiology of Parkinson's Disease. In: Movement Disorders: Neurologic Principles and Practice, 2nd, Watts, RL, Koller, WC (Eds), The McGraw-Hill Companies, New York p Hornykiewicz O. The discovery of dopamine deficiency in the parkinsonian brain. J Neural Transm Suppl 2006; :9. 4. Olanow CW, Watts RL, Koller WC. An algorithm (decision tree) for the management of Parkinson's disease (2001): treatment guidelines. Neurology 2001; 56:S1. 5. Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA 2014; 311:1670
, The McGraw-Hill Companies, New York p Hornykiewicz O. The discovery of dopamine deficiency in the parkinsonian brain. J Neural Transm Suppl 2006; :9. 4. Olanow CW, Watts RL, Koller WC. An algorithm (decision tree) for the management of Parkinson s disease (2001): treatment guidelines. Neurology 2001; 56:S1. 5. Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: a review. JAMA 2014; 311:1670")
Apresentações semelhantes
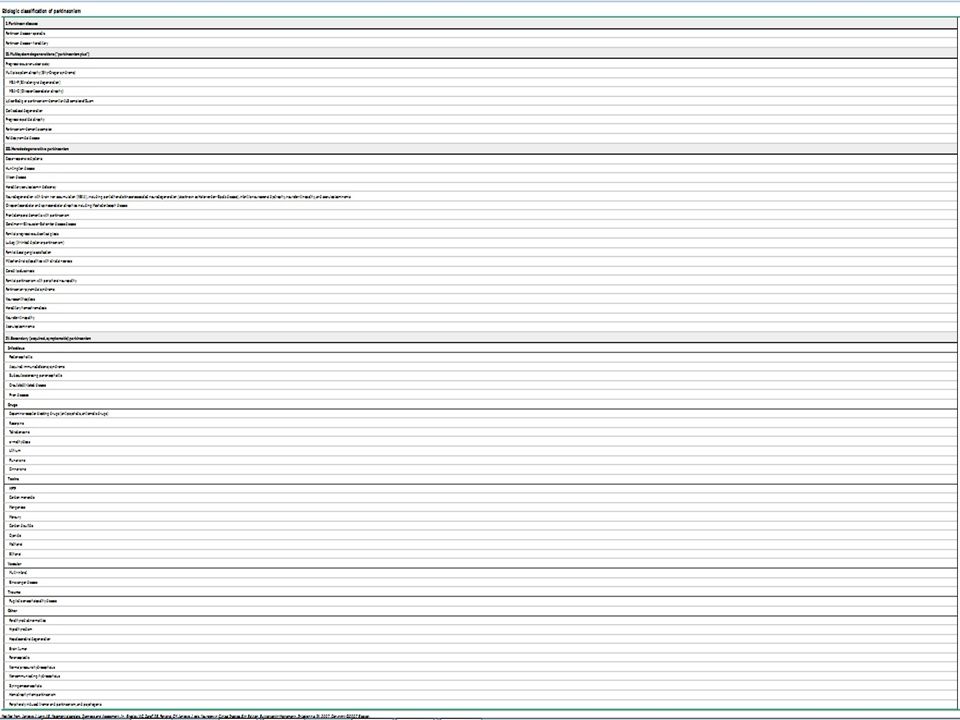






, e que pode levar a incapacitação funcional.>")












