Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Caso Clinico: Imunodeficiência combinada severa (SCID)
Danillo Hellou Coordenação:Dr. Paulo R. Margotto Dra. Fabíola Scancetti Tavares Medicina – ESCS/SES/DF

2
Identificação RN de O.R.M.L de 42 anos Sexo feminino 1h de vida
Mãe procedente de Braslândia-GO

3
História Clínica RN deu entrada ao HRAS com uma hora de vida, após parto normal em automóvel da família. Trata-se de terceiro filho de casal consangüíneo (primos de primeiro grau). Veio para investigação devido a possibilidade de apresentar imunodeficiência primária.
. Veio para investigação devido a possibilidade de apresentar imunodeficiência primária.")
4
História Clínica Primeiro filho do casal falecido aos seis meses de idade por pneumonia e septicemia (sic). Segundo filho do casal nasceu bem.Aos dois meses, após vacinação contra BCG apresentou supuração no local além de pneumonia e monilíase oral e perineal. Recebeu alta hospitalar com esquema tríplice e voltou após dois meses com a mesma pneumonia.Nesta ocasião foi revelado leucopenia severa e hipogamaglobulinemia quando foi diagnosticado Imunodeficiência combinada severa (SCID: se lê “squid”).
.")
5
Antecedentes Fisiológicos
Mãe 42 anos. G3P3A0 PN3 Realizou 6 consultas pré-natal. Gestação sem intercorrências. Nasceu com 38 sem + 2 dias. PN no automóvel. Peso 3320g PC: 34cm Est: 50cm Apgar de 8 e 10 em 1° e 5° min. Amamentando ao seio materno.

6
Antecedentes familiares
Pai e mãe consangüíneos: primos de 1° grau. Irmão: falecido aos 6 meses com pneumonia e septicemia(sic). Irmão: falecido aos 8 meses por pneumonia. Portador de SCID. Meio irmã: saudável. Não há relatos de outras mal formações ou doenças genéticas na família.
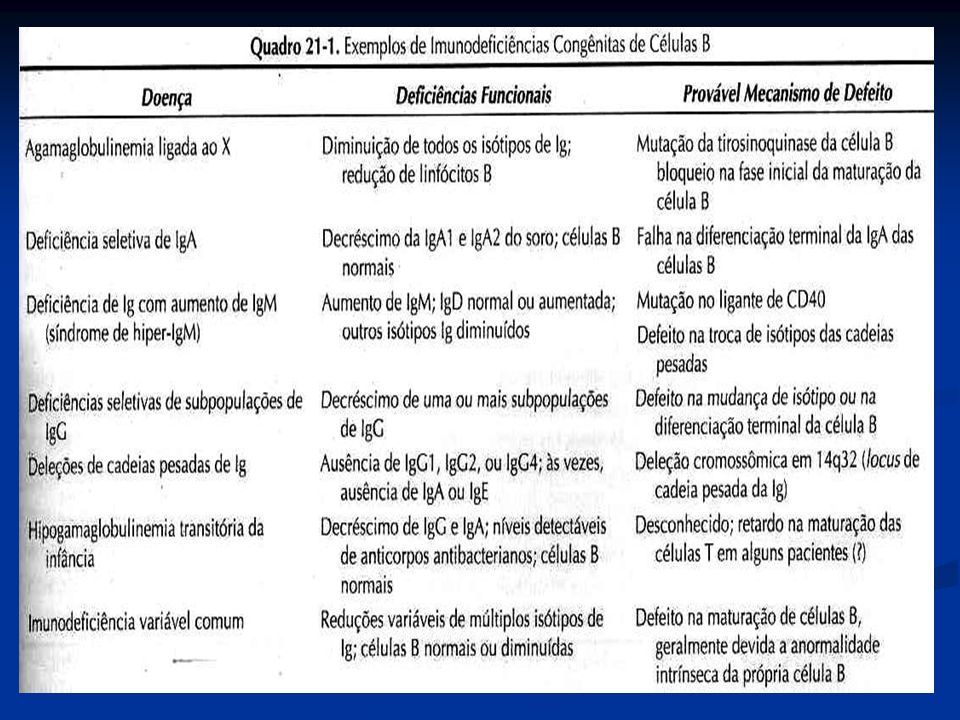
. Irmão: falecido aos 8 meses por pneumonia. Portador de SCID. Meio irmã: saudável. Não há relatos de outras mal formações ou doenças genéticas na família.")
7
Exame Físico BEG, ativa e reativa, hidratada, corada, acianótica, anictérica, eupneica, afebril. AR: MVF s/ RA. ACV: RCR 2T, BNF s/ SOPRO. ABD: Sem Alterações. EXT: perfundidas sem edema. Não foi notada nenhuma anormalidade ou mal-formação no exame físico.

8
Evolução Paciente permaneceu em isolamento de contato, com boa evolução clinica. Apresentou icterícia no terceiro dia de vida +/4+, zona II de Kramer com boa resposta à fototerapia azul. Evoluiu sem intercorrências com alta hospitalar no décimo dia de internação.

9
Exames Complementares

10
Exames Complementares
Dia 1° dia 2° dia 3° dia Leucócitos 19700 9100 7500 HG 13.3 17.6 18.4 HT 40.1% 53.0% 55.1% Plaq 87.000

11
Exames Complementares 3° dia
IgG IgA IgM Resultado 1850 < 25.1* < 17.4* Valor Ref 4 - 27 *valor mínimo detectável pelo método usado

12
Exames Complementares
Hemocultura: após 7 dias da colheta que foi no 2° dia de vida do paciente foi negativa. Mesmo assim decidido continuar antibioticoterapia (ATB) para germes do canal do parto até completar 10 dias de ATB.
 para germes do canal do parto até completar 10 dias de ATB.")
13
Exames Complementares
Bilirrubinemia: : BT 20.2 BI 19.6 BD 0.6 : BT 14.6 BI 13.4 BD 1.2 Coombs Direto negativo TS: O positivo

14
Diagnostico Enviado amostra de sangue para Imunologia Pediátrica UNIFESP Baixíssimo n° de linfócitos. Pela história e exames complementares. Diagnosticado Imunodeficiência Combinada Severa.(SCID)
")
15
Tratamento Realizado Profilaxia: Terapêutico: Isolamento de contato
Antibioticoterapia (ampicilina, gentamicina, bactrim em doses terapêuticas) Nistatina VO Contra-indicado vacinação. (BCG) Terapêutico: Fototerapia Dupla Azul. Indicado infusão de IgG de 21 em 21 dias. Encaminhada para ambulatório do Hospital de Base de Brasília
 Nistatina VO. Contra-indicado vacinação. (BCG) Terapêutico: Fototerapia Dupla Azul. Indicado infusão de IgG de 21 em 21 dias. Encaminhada para ambulatório do Hospital de Base de Brasília.")
16
Imunodeficiências Primárias
DISCUSSÃO Imunodeficiências Primárias

17
Introdução Integridade do sistema imune é essencial para defesa contra microorganismos e seus produtos tóxicos. Mecanismo complexo e interdependente. Falha resulta em infecções graves e refratárias. Predisposição para câncer.

18
Classificação das ID Classificadas em primárias ou congênitas e secundárias ou adquiridas. As primárias são defeitos gênicos que se expressam na primeira ou segunda infância. As secundárias são de origem nutricional, neoplásica, uso de quimioterápico ou infecção (HIV).
.")
19
Características Gerais
Suceptibilidade a infecções. Tipo de infecções depende do tipo de defeito. Humoral Piogênicas. Celular Vírus e bactérias intracelulares. Tendência aumentada para cânceres. Clinica e patologicamente heterogêneas.

20
Defeitos primários da células B
Resultam em deficiente produção de anticorpos. Resposta Celular preservada. Infecções recorrentes por bactérias piogênicas encapsuladas: pneumococo, estreptococo, meningococo e Haemofilus influenzae. Poliomielite por Vacina. Parasitoses intestinais: Giardíase.

21
Defeitos primários da células B
A anormalidade encontra-se em diferentes estágios da diferenciação do linfócito B. Pode ser apenas funcional. Podem ser: Agamaglobulinemia ligada ao X, Deficiências seletivas de Ig, Imunodeficiência comum variável. Injeções periódicas de Ig ( Imunidade passiva).
.")
23
Defeitos Primários nas células T
Induz a perturbação das reações imunes celulares. Afetam Diferenciação/Maturação de células T. Aumenta a suscetibilidade a infecções por vírus, fungos, bactérias intracelulares e protozoários. Aumento de incidência de Neoplasias.

24
Defeitos Primários nas células T
Como resultado, estas infecções são graves e difícil controle. Normalmente indivíduos afetados não chegam a idade adulta. Principais patógenos envolvidos: Cândida, P. carinii, Varicela.

25
Defeitos Primários nas células T
Resposta anormalmente baixa a estimuladores policlonais como a PHA. Induzem também perturbação na imunidade Humoral já que as células T auxiliam na apresentação de antígenos. Principais exemplos: Síndrome de DiGeorge, Síndrome de Wiskott-Aldrich.

26
Imunodeficiências Combinadas Severas (SCID)
Grupo heterogêneo de distúrbios. Caracteriza-se por anormalidade de desenvolvimento ou função de linfócitos. Deficiência da imunidade Celular e humoral. Linfocitopenia profunda associada a hipoplasia tímica e tecidos linfóides, e deficiência de Ig.

27
SCID Manifesta-se usualmente no primeiro ano de vida.
Mais comumente : Candidíase oral, Diarréia crônica infecciosa, desenvolvimento retardado. Por fim infecções por qualquer microorganismo com alta gravidade.

28
SCID Transmissão em 40% das vezes é autossômica recessiva.
Metade desses o defeito genético não é conhecido. Suspeita-se de defeito da expressão do gene de receptores antigênicos (receptores de superfície).
.")
29
SCID Em 50% das SCID autossômicas recessivas o defeito é no gene ADA (desaminase da adenosina) do cromossomo 2. Toxicidade resultando em defeito na replicação do DNA. Reduzido número de linfócitos B e T.

30
Genes Anormais em SCID Buckley RH, 2004

31
Frequência relativa dos diferentes tipos genéticos de SCID entre 160 pacientes
Relative frequencies of the different genetic types of SCID among 160 patients seen consecutively over 3 decades. RD, Reticular dysgenesis; CHH, cartilage hair hypoplasia. Buckley HR,2002

32
SCID 60% das SCID são de origem recessiva ligada ao X.
Mutação do gene “Gama Comum” que codifica uma cadeia de aa compartilhada pelos receptores de interleucina 2, 4, 7, 9 e 15. Perturbação na diferenciação de linfócitos T. Cels B aumentadas porém não funcionantes.

33
SCID

34
SCID

35
SCID Buckley RH, 2004

36
SCID Rx de tórax mostrando campos pulmonares normais e
sombra tímica normal Buckley RH, 2006

37
SCID Buckley RH, 2004

38
SCID Buckley RH, 2004

39
Tratamento SCID Trata-se de uma emergência pediátrica.
ATB profilática e se necessário terapêutica. Transplante de medula com até 95% de cura se realizado antes do terceiro mês de vida. Observar a doença enxerto versus hospedeiro.

40
SCID Buckley RH, 2004

41
Caso clínico: Imunodeficiências primárias
Consultem: Caso clínico: Imunodeficiências primárias Autor(es): Lenira Silva Valadão, Elisa de Carvalho
: Lenira Silva Valadão, Elisa de Carvalho.")
42
Ddo Danilo Dr. Paulo R. Margotto Dra. Fabíola Scancetti Tavares OBRIGADO! 13/4/2007

Apresentações semelhantes