Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Rafael Moreno F. de Araújo
Síndrome de Down Maycow A. Patrício Rafael Moreno F. de Araújo Rafael Oppermann Rafael T. Cardoso Roberto T. Sant’Anna Vinícius S. S. Oliveira

2
Introdução A Síndrome de Down ou trissomia do cromossomo 21 é o distúrbio cromossômico mais comum.

3
Histórico 1866 - John Langdon Haydon Down 1930 - Jenkins e Penrose
Waardenburg e Bleyer Lejeune Clark O primeiro diagnóstico pré-natal 1983 em diante - AFP, HCG, estriol-não conjugado, outros

4
Epidemiologia A freqüência cumulativa de nascimentos com trissomia do 21, 18, 13 é menor que 1%; A taxa de nascimentos com Síndrome de Down é de 1:700.

5
Prevalência de acordo com a idade materna:
Epidemiologia Prevalência de acordo com a idade materna: 20-24 anos - 1:1400 acima de 35 anos - 1:350 acima de 45 anos - 1:25

6
A freqüência em relação à idade materna possui basicamente 2 tipos de distribuição:

7
Inglaterra X Singapura
As contradições: Inglaterra X Singapura

8
Síndrome de Down - Etiologia
Introdução: Causa exata ainda não descoberta. Possível causa relacionada à idade materna avançada. Três formas de alterações cromossômicas: trissomia livre do 21, translocação cromossômica e mosaicismo (trissomia livre parcial).
.")
9
Síndrome de Down - Etiologia
2. Trissomia do cromossomo 21: 95% dos portadores de síndrome de Down (47,XX,+21; 47,XY,+21). Frequentes em progênies de mães com idade avançada: erros na divisão meiótica e mecanismos de não-disjunção cromossômica (Warburton et al). Fatores ambientais ou externos relacionados, mas não confirmados (Hook, 1992). Não-disjunção do cromossomo 21 (mecanismo genético) gameta com 24 cromossomos. Erros na primeira divisão meiótica: 90% por não-disjunção na meiose I materna (Petersen et al.,1993)
. Frequentes em progênies de mães com idade avançada: erros na divisão meiótica e mecanismos de não-disjunção cromossômica (Warburton et al). Fatores ambientais ou externos relacionados, mas não confirmados (Hook, 1992). Não-disjunção do cromossomo 21 (mecanismo genético) gameta com 24 cromossomos. Erros na primeira divisão meiótica: 90% por não-disjunção na meiose I materna (Petersen et al.,1993)")
10
Síndrome de Down - Etiologia
5% cromossomo de origem paterna: eventos não-disjuncionais na meiose II mais frequentes (2x). Outro mecanismo citogenético relacionando idade materna avançada e redução do comprimento 21q (Sherman et al., 1994).
. Outro mecanismo citogenético relacionando idade materna avançada e redução do comprimento 21q (Sherman et al., 1994).")
11
Síndrome de Down – Trissomia 21

12
Síndrome de Down - Etiologia
3. Síndrome de Down por translocação: Corresponde a cerca de 3-4% dos casos de síndrome de Down. Cromossomo 21 fusionado a um cromossomo acrocêntrico. Número de cromossomos normal, porém há triplicação do material do cromossomo 21. Cariótipo com 46 cromossomos e a translocação é representada como t(14;21) ou t(14q21q). Translocações balanceadas: não há excesso ou perda de material genético (carreador balanceado).
 ou t(14q21q). Translocações balanceadas: não há excesso ou perda de material genético (carreador balanceado).")
13
Síndrome de Down - Etiologia
Translocações podem ser herdadas análise dos cromossomos parentais (possível carreador). Translocações com cromossomos do grupo D: 40% herdados (90% destes a mãe é carreadora). Somente 7% das translocações com cromossomos do grupo G têm carreador parental (Hook,1981). Translocações de novo t(21;21): fusão de cromátides irmãs (isocromossomos) iso(21q) (Shaffer,1993). Origem em células germinativas maternas, em cromossomos que não estão em fusão cêntrica ou braços cromossômicos trocados (Petersen,1991).
. Translocações com cromossomos do grupo D: 40% herdados (90% destes a mãe é carreadora). Somente 7% das translocações com cromossomos do grupo G têm carreador parental (Hook,1981). Translocações de novo t(21;21): fusão de cromátides irmãs (isocromossomos) iso(21q) (Shaffer,1993). Origem em células germinativas maternas, em cromossomos que não estão em fusão cêntrica ou braços cromossômicos trocados (Petersen,1991).")
14
Síndrome de Down - Translocação

15
Translocação robertsoniana balanceada

16
Síndrome de Down - Etiologia
4. Mosaicismo do cromossomo 21: Presença de duas ou mais populações de células que diferem no seu conteúdo cromossômico. Decorrem da não-disjunção ou do atraso anafásico durante as primeiras divisões mitóticas embrionárias. Obtenção de linhagens com cariótipo 47,+21 e outras com cariótipo normal. Mosaicismo celular: mistura de linhagens de um mesmo tipo celular. Mosaicismo tecidual: um tipo celular normal e outro grupo celular com trissomia do 21.

17
Síndrome de Down - Etiologia
Número de células trissômicas X envolvimento fenotípico. Em 20% dos casos o cromossomo extra provém de não-disjunção mitótica em embrião diplóide após fertilização (Richards,1974). Linhagens celulares com cariótipo 47,+21 sobrevivem; linhagens com cariótipo 45,-21 morrem embrião com linhagens celulares normais e trissômicas. Pangalos et al. (1994): erros meióticos que levariam a trissomia do 21 em 10/17 casos estudados.
. Linhagens celulares com cariótipo 47,+21 sobrevivem; linhagens com cariótipo 45,-21 morrem embrião com linhagens celulares normais e trissômicas. Pangalos et al. (1994): erros meióticos que levariam a trissomia do 21 em 10/17 casos estudados.")
18
Síndrome de Down 5. Risco de recorrência: Depende da etiologia.
Trissomia livre do cromossomo 21: risco estimado em 1% para casal que já tem um filho afetado. Hook et al. (1992): risco 1% maior que o risco para a idade específica risco para mães 35 anos em relação àquelas com mais de 35 anos. Taxa de recorrência influenciada pela idade materna na primeira gestação 1º gestação antes dos 30 anos tem maior risco.
: risco 1% maior que o risco para a idade específica risco para mães 35 anos em relação àquelas com mais de 35 anos. Taxa de recorrência influenciada pela idade materna na primeira gestação 1º gestação antes dos 30 anos tem maior risco.")
19
Síndrome de Down Translocação: depende de qual genitor provém a alteração cromossômica. Mãe (carreadora balanceada): 10-15% de risco. Pai (carreador balanceado): 5% de chance. Cerca de ¾ dos casos nenhum genitor é o carreador (mutação em células germinais de um deles). Translocação t(21;21): 100% de chance de recorrência quando um genitor é carreador balanceado. Mosaicismo: em torno de 3%. Sachs (1990): mosaicismo gonadal como causa de risco de recorrência elevado.
: 5% de chance. Cerca de ¾ dos casos nenhum genitor é o carreador (mutação em células germinais de um deles). Translocação t(21;21): 100% de chance de recorrência quando um genitor é carreador balanceado. Mosaicismo: em torno de 3%. Sachs (1990): mosaicismo gonadal como causa de risco de recorrência elevado.")
20
MAPEAMENTO DO CROMOSSOMO 21

21
ASPECTOS GERAIS O cromossomo 21 é o menor autossomo humano.
Uma cópia extra do cromossomo 21 causa a Síndrome de Down. Alguns loci anônimos para desordens monogênicas e predisposições para desordens complexas comuns também foram mapeadas neste cromossomo A perda da heterozigoticidade foram observadas em regiões associadas a tumores sólidos. Foram seqüenciados pares de bases (pb) de DNA, sendo que o maior seqüência contígua possui pb. Apenas três pequenos clones de intervalos e sete seqüências de intervalos restaram, compreendendo em torno de 100 kilobases. Foram alcançados 99,7% de segurança de 21q. Também foram seqüenciados bp do braço curto. Os aspectos estruturais identificados incluem duplicações que provavelmente envolvidos em anormalidades cromossômicas e estruturas repetidas nas regiões teloméricas e pericentroméricas. A análise do cromossomo revelou 127 genes conhecidos, 98 genes previstos e 59 pseudogenes
 de DNA, sendo que o maior seqüência contígua possui pb. Apenas três pequenos clones de intervalos e sete seqüências de intervalos restaram, compreendendo em torno de 100 kilobases. Foram alcançados 99,7% de segurança de 21q. Também foram seqüenciados bp do braço curto. Os aspectos estruturais identificados incluem duplicações que provavelmente envolvidos em anormalidades cromossômicas e estruturas repetidas nas regiões teloméricas e pericentroméricas. A análise do cromossomo revelou 127 genes conhecidos, 98 genes previstos e 59 pseudogenes.")
22
ASPECTOS GERAIS Cromossomo 21 representa cerca 1-1,5% do genoma humano. O cromossomo 21 foi o primeiro autossomo que foram desenvolvidos um mapa de acoplamento denso, um mapa físico do cromossomo artificial de levedura (YAC) e um mapa de restrição. O tamanho do braço longo do cromossomo (21q) foi estimado para ter cerca de 38 megabases (Mb), baseado em estudos por PFGE (pulsed-field gel electrophoresis) usando fragmentos de restrição. Desde de 1995, quando o seqüenciamento foi iniciado, cerca de 60 RNAs mensageiros específicos do cromossomo 21 foram caracterizados.
 e um mapa de restrição. O tamanho do braço longo do cromossomo (21q) foi estimado para ter cerca de 38 megabases (Mb), baseado em estudos por PFGE (pulsed-field gel electrophoresis) usando fragmentos de restrição. Desde de 1995, quando o seqüenciamento foi iniciado, cerca de 60 RNAs mensageiros específicos do cromossomo 21 foram caracterizados.")
23
CATÁLAGO GENÔMICO A geografia do cromossomo foi feita através de mapeamento e seqüenciamento resultando em um catálogo genômico. O catálogo dos genes do cromossomo 21 contém: genes conhecidos, genes previstos in silico através da análise da seqüencia genômica e pseudogenes. O catálogo é dividido em cinco categorias hierárquicas principais para distinguir genes conhecidos dos puramente previstos, e ainda seqüências anônimas complementares de DNA das que exibem similaridades para reconhecer proteínas ou domínios moduladores. O critério usado para a classificação dos genes foi baseado em resultados de integrados resultados de análises computadorizadas usando programas de previsão de exon e procura de similaridades seqüenciais. Foram usados os seguintes parâmetros: (1) Exons codificadores facultativos foram predizidos usando programas GRAIL, GENSCAN e MZEF (2) Seqüências de nucleotídios identificados foram usados para expressar seqüências de TAGS (ESTs) (3) Similaridades dos aminoácidos para reconhecer proteínas ou moduladores dos domínios funcionais foram considerados significantes quando a identidade total era maior que 25% quando mais de 50 resíduos de aminoácidos foram observados.
 Exons codificadores facultativos foram predizidos usando programas GRAIL, GENSCAN e MZEF. (2) Seqüências de nucleotídios identificados foram usados para expressar seqüências de TAGS (ESTs). (3) Similaridades dos aminoácidos para reconhecer proteínas ou moduladores dos domínios funcionais foram considerados significantes quando a identidade total era maior que 25% quando mais de 50 resíduos de aminoácidos foram observados.")
24
CATEGORIAS DOS GENES O cromossomo contém 225 genes e 59 pseudogenes.
127 correspondem a genes conhecidos (subcategorias 1.1 e 1.2) 98 representam novos genes facultativos previstos in silico (subcategorias 2.1 e 2.2) 17 são anônimos ORFs caracterizando domínios modulares (subcategorias 3.1 e 3.2) A maior parte (68 genes) são unidades de transcrição anônimas sem nenhuma similaridade com proteínas conhecidas (subcategorias 4.1, 4.2 e 4.3). Dados mostram que cerca de 41% dos genes que foram identificados no cromossomo 21 não têm atributos funcionais.
 98 representam novos genes facultativos previstos in silico (subcategorias 2.1 e 2.2) 17 são anônimos ORFs caracterizando domínios modulares (subcategorias 3.1 e 3.2) A maior parte (68 genes) são unidades de transcrição anônimas sem nenhuma similaridade com proteínas conhecidas (subcategorias 4.1, 4.2 e 4.3). Dados mostram que cerca de 41% dos genes que foram identificados no cromossomo 21 não têm atributos funcionais.")
25
COMPARAÇÃO COM OUTROS AUTOSSOMOS
EM RELAÇÃO AO CROMOSSOMO 22: Ambos são acrocêntricos e têm tamanhos similares. São detectados em ambos cromossomos seqüências parecidas nas regiões pericentroméricas e sub-teloméricas. A densidade gênica é maior no cromossomo 22. EM RELAÇÃO A OUTROS AUTOSSOMOS: Detecta-se que seqüências repetidas similares a 93-bp do cromossomo 21 são encontradas nos cromossomos 22, 10 e 19. Também são detectados alguns regiões parálogas entre o cromossomo 21 e outros cromossomos humanos.

26
IMPLICAÇÕES MÉDICAS Síndrome de Down Neoplasias
Desordens monogênicas: uma forma de Doença de Alzheimer, esclerose lateral amiotrófica, doença poliglandular autoimune, homocistinúria e epilepsia mioclonal progressiva, predisposição à leucemia. Doenças complexas: distúrbio bipolar afetivo e uma hiperlipidemia familiar

27
SÍNDROME DE DOWN Resultado da presença de três cópias do cromossomo 21 ao invés de duas. Dados colhidos de ratos transgênicos indicam que apenas um subgrupo de genes do cromossomo 21 pode estar envolvido com os fenótipos da Síndrome de Down. Apesar de ser difícil de selecionar genes candidatos para estes fenótipos, alguns produtos gênicos podem ser mais sensíveis ao desequilíbrio da dose gênica do que outros. Nestes podem-se incluir morfogenes, moléculas de adesão às células, componentes de multi-subunidades proteicas, ligantes e seus receptores, reguladores de transcrição e transportadores.

28
QUAIS GENES ESTÃO ENVOLVIDOS ?
Só uma pequena porção do cromossomo 21 precisa triplicada para causar as alterações vistas na síndrome de Down ; esta é chamada REGIÃO CRÍTICA Contudo ela não é uma porção isolada, mas possivelmente várias áreas que não estão necessariamente lado a lado . Estima-se que 20 a 50 genes dos 200 a 250 presentes no cromossomo façam parte da região crítica (não se sabe ao certo quais) .
 .")
29
SÍNDROME DE DOWN Genes em que sua superexpressão podem estar relacionados a Síndrome de Down: · Superóxido Dismutase (SOD1) – superexpressão pode causar envelhecimento precoce e decréscimo da função do sistema imune, que podem estar relacionados com a Demência Senil do Alzheimer. · COL6A1 – superexpressão pode causar defeitos cardíacos · ETS2 – superexpressão pode causar anormalidades esqueléticas e/ou leucemia · CAF1A – superexpressão pode prejudicar a síntese de DNA · Cistatione Beta Sintase (CBS) – superexpressão pode romper o metabolismo e reparo ao DNA · DYRK – superexpressão pode causar retardo mental · CRYA1 – superexpressão pode causar catarata · GART – superexpressão pode romper a síntese e reparo do DNA · IFNAR – gene da expressão do Interferon, sua superexpressão pode interferir no sistema imune e em outros sistemas orgânicos · Proteína Beta Amilóide – maior componente das placas de neurofilamentos, pode estar relacionado à patogênese do Alzheimer
 – superexpressão pode causar envelhecimento precoce e decréscimo da função do sistema imune, que podem estar relacionados com a Demência Senil do Alzheimer. · COL6A1 – superexpressão pode causar defeitos cardíacos. · ETS2 – superexpressão pode causar anormalidades esqueléticas e/ou leucemia. · CAF1A – superexpressão pode prejudicar a síntese de DNA. · Cistatione Beta Sintase (CBS) – superexpressão pode romper o metabolismo e reparo ao DNA. · DYRK – superexpressão pode causar retardo mental. · CRYA1 – superexpressão pode causar catarata. · GART – superexpressão pode romper a síntese e reparo do DNA. · IFNAR – gene da expressão do Interferon, sua superexpressão pode interferir no sistema imune e em outros sistemas orgânicos. · Proteína Beta Amilóide – maior componente das placas de neurofilamentos, pode estar relacionado à patogênese do Alzheimer.")
30
Mapa fenotípico do cromossomo 21

31
NEOPLASIAS Neoplasias estão relacionadas com a perda da heterozigoticidade observadas em regiões específicas do cromossomo 21. Alguns tumores sólidos incluindo cânceres de cabeça e pescoço, mama, pâncreas, boca, estômago, esôfago e pulmão. A perda da heterozigoticidade indica que deve existir pelo menos um gene supressor de tumor neste cromossomo. A incidência baixa de tumores sólidos em indivíduos com Síndrome de Down indica que uma dose aumentada de alguns genes do cromossomo 21 pode proteger estes indivíduos destes tumores De outro modo, pacientes com Síndrome de Down têm um aumento marcado na incidência de leucemias na infância, e a trissomia do cromossomo 21 em células blásticas é uma das mais comuns aneuploidias cromossômicas vistas em leucemias de crianças.

32
Aspectos Clínicos

33
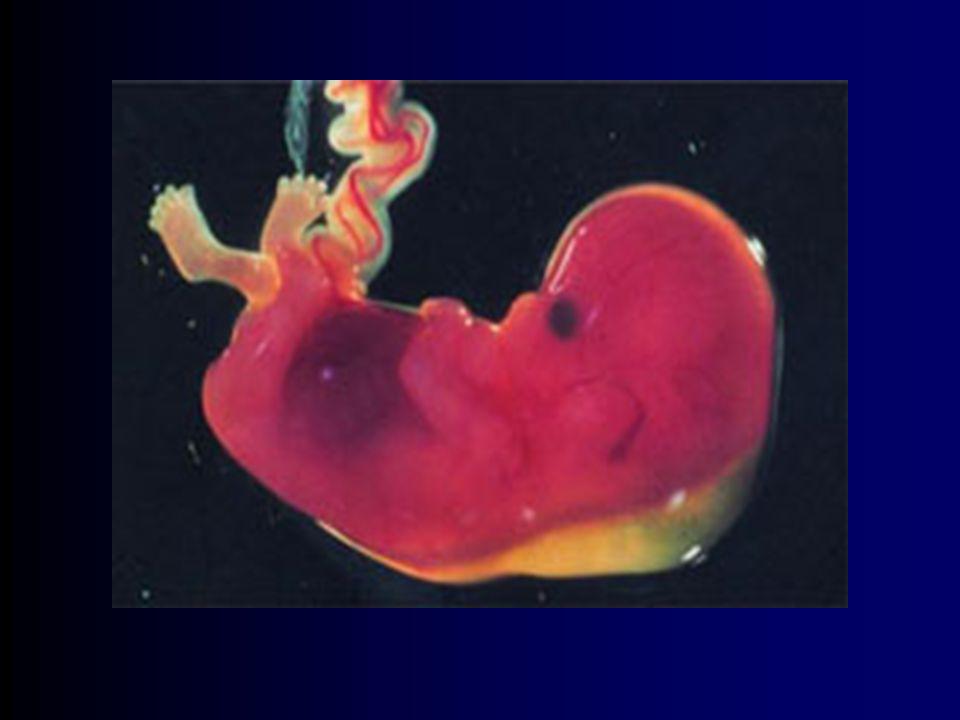
Aspectos Fetais Características mais prevalentes linha siamesa palmar
clinodactilia defeitos cardíacos septais feto de menor tamanho espessamento nucal fácies não patognomônica

36
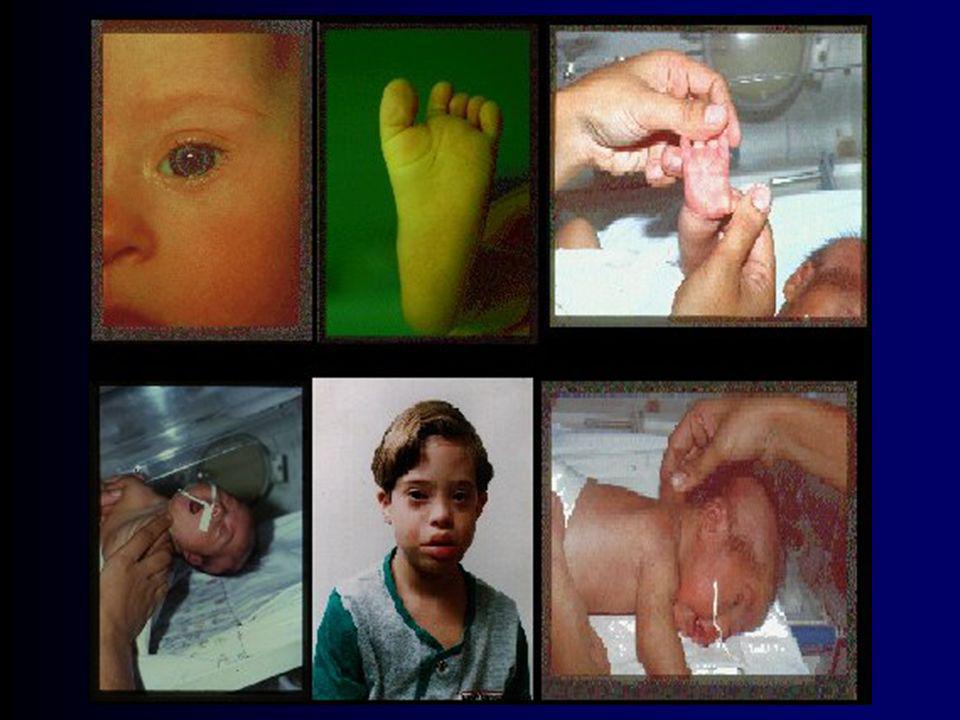
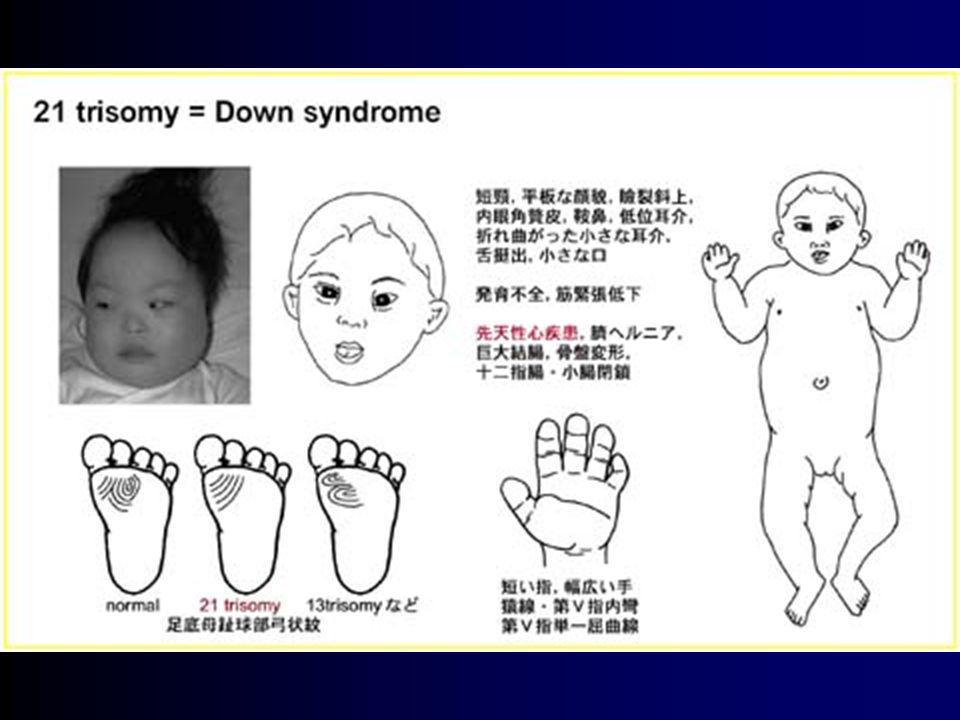
Aspectos da Infância Características mais prevalentes:
fácies mongol (Gestalt face) encéfalo ovalado/terceira fontanela relativa hipotonia olhos “chorosos” pescoço encurtado linha palmar solitária glossoprotrusão
 encéfalo ovalado/terceira fontanela. relativa hipotonia. olhos chorosos pescoço encurtado. linha palmar solitária. glossoprotrusão.")
37
Aspectos da Infância Aspectos menos prevalentes: braquicefalia
base nasal alargada clinodactilia do quinto digital linha plantar entre 1 e 2 dedos com grande espaçamento interdigital quadros leucemóides *alta variabilidade.

40
Índice Diagnóstico em Neonatos

41
Aspectos Clínicos no Adulto
Semelhantes aos da infância, mas com alterações associadas ao crescimento e puberdade. Aspectos mais prevalentes: expressão dos defeitos cardíacos distúrbios gastrointestinais alterações de ordem cognitiva e comportamental não coerentes com idade cronológica

42
Clínica do adulto Dificuldade em manter um peso adequado
Distúrbios das vias aéreas superiores e trato respiratório recorrentes que compreendem alterações de ordem histológica e mecânica e que aumentam a morbidade pós tratamentos de correção.

43
Aspectos do mosaicismo do cromossomo 21
relação entre linhagens diploidiais e aneuploidiais detecção difícil que em alguns casos pede avaliação cariotípica impossibilidade de predição mesmo com estudos de proporcionalidade entre linhagens celulares

44
Crescimento e Desenvolvimento
Ao nascer apresenta discreto retardo no crescimento, peso, altura e diâmetro cefálico na média. Velocidade de crescimento normal até o 1 ano de vida aonde reduz-se voltando a um novo aumento aos 3 anos. A partir dos 3 anos normaliza-se e tende a não cair mais. Puberdade ocorre normalmente apesar de alguns indivíduos hipogonádicos. O pico puberal é restrito e altura final acaba em 140 a 160 cm. (IGF1 insuficiente, GH é efetivo para 95 em percentil)
")
45
Down e Obesidade Tendência ao sobrepeso já na primeira infância.
Freqüente no indivíduo adulto Prevalência aproximada de 96% homens e 71% mulheres* Prevenção suplanta regimes drásticos (características metabólicas próprias de economia energética).
.")
46
Desenvolvimento Intelectual e da Personalidade
Alterações cerebrais na síndrome Down: maturação deficiente dos dendritos atrofia e deposição precoce de material amilóide no tecido nervoso central Desenvolvimento “ilimitado” associado ao grau de estimulação Personalidade multivariada. Maior susceptibilidade a desordens de conduta e psicose

47
Desenvolvimento cognitivo, linguístico e motor
Alterações emocionais e de relação são praticamente imperceptíveis no 1 ano de vida. Alterações motoras e de linguagem já se tornam aparentes em torno do segundo ano de vida. Hipotonia, retardo mental e déficit motor variáveis

48
Desenvolvimento cognitivo, linguístico e motor
QI entre 20 e 85 Períodos de start/stop nas esferas cognitiva, linguística e motora: 0 a 4 anos - aceleração1 (i.m.f. 17meses) 5 a 9 anos - aceleração2 (i.m.f. 31meses) 8 a 11 anos - platô 12 a 13 anos - aceleração3 (i.m.f 40 meses) possibilidade de i.m.f 90 a 100 meses se submetido a estímulo apropriado considerar fatores epigenéticos.
 5 a 9 anos - aceleração2 (i.m.f. 31meses) 8 a 11 anos - platô. 12 a 13 anos - aceleração3 (i.m.f 40 meses) possibilidade de i.m.f 90 a 100 meses se submetido a estímulo apropriado. considerar fatores epigenéticos.")
51
Desenvolvimento sexual Masculino
Mesmos atributos dos não sindrômicos Fertilidade discutida (somente um caso conhecido no mundo) Excesso de peso e baixa estatura influenciam comportamento sexual. Puberdade inicia-se em torno dos 13 e completa-se aos 17 anos em média. Poucos chegam ao auge das características sexuais secundárias Controvérsia em relação ao desenvolvimento genital e testicular.
 Excesso de peso e baixa estatura influenciam comportamento sexual. Puberdade inicia-se em torno dos 13 e completa-se aos 17 anos em média. Poucos chegam ao auge das características sexuais secundárias. Controvérsia em relação ao desenvolvimento genital e testicular.")
52
Sexualidade Masculina
Apresentam alterações hormonais nos níveis de FSH/LH o que supõe uma disfunção primária das glândulas sexuais Observação não rara de oligospermia e azoospermia na síndrome de Down. Dados porém inconclusivos.

53
Desenvolvimento Sexual Feminino
Raramente se observam alterações em genitais externos Menarca entre 11 e 13 anos em média TPM É subfértil (algumas não avulam e outras apresentam alterações na ovulação) Dos casos de reprodução observados, têm-se em torno de 30% de conceptos com Down (apesar do risco teórico de 50%), sendo que destes, aproximadamente 10% resultam e aborto e quase 60% vêm a termo sem a síndrome. Das crianças que nasceram sem a síndrome, em torno de 20% apresentaram algum comprometimento físico ou mental (incesto ou problemas no parto)
 Dos casos de reprodução observados, têm-se em torno de 30% de conceptos com Down (apesar do risco teórico de 50%), sendo que destes, aproximadamente 10% resultam e aborto e quase 60% vêm a termo sem a síndrome. Das crianças que nasceram sem a síndrome, em torno de 20% apresentaram algum comprometimento físico ou mental (incesto ou problemas no parto)")
54
Sexualidade Vida sexual ativa é incomum
Vida social tende a ser restrita e quando há relacionamento com o sexo oposto é mais de amizade que de namoro propriamente dito Não há provas quanto a uma suposta hipersexualidade. Manifestações exageradas tendem a ser fruto do déficit mental mais do que de um impulso sexual extremo.

55
Alterações mais comuns na clínica do paciente com Down
Doença Cardíaca Congênita prevalência de malformações entre 16 e 62% defeitos mais comuns: alterações atrioventriculares com disfunção septal, Tetralogia de Fallot Se não tratados têm mortalidade em torno de 50%. As morbidades do paciente adulto se correlacionam basicamente à hipertensão pulmonar e falência do coração direito.

56
Malformação Átrio-ventricular congênita na S. de Down

57
Alterações Gastrointestinais
Atresia duodenal (2 a 5%) D. de Hirschsprung (2%) evolui para enterocolite Onfalocele, pâncreas em anel, malformação anorretal. Doença Celíaca? Constipação com origem na hipotonia colônica e fatores dietéticos
 D. de Hirschsprung (2%) evolui para enterocolite. Onfalocele, pâncreas em anel, malformação anorretal. Doença Celíaca Constipação com origem na hipotonia colônica e fatores dietéticos.")
58
Outras Alterações Oftalmológicas:
catarata congênita, estrabismo, glaucoma e erros refracionários maiores Pobre acuidade visual Prevalência em torno de 60% Aditivas: malformação das estruturas do ouvido médio. Prevalência entre 40 e 75% Infecções comuns por malformação anatômica dos dutos

59
Sistema Imune em Geral Suceptibilidade aumentada à infecção como causa principal de morbimortalidade Ausência de imunodeficiência franca, mas com sistema defectivo. Microcitose e leucopenia já na vida fetal.

60
Imunidade Celular Timo reduzido com menos córtex e depleção de timócitos. Expansão defeituosa de precursores de células T e células NK defeituosas o que leva a um pool celular incompleto e resposta celular anormal. Baixos níveis de fitohemaglutinina e IL’s. Redução da capacidade proliferativa em resposta a antígenos e mitógenos.

61
Imunidade Humoral Bem menos afetada do que a imunidade celular
A resposta de anticorpos a vários antígenos e proteínas virais é pobre A prevalência de HbsAg em Down é maior que na população em geral, respondem menos à vacina contra HBV.

62
Imunidade Inespecífica
O número de polimorfonucleares é normal, mas há importante deficiência na quimiotaxia dos neutrófilos, baixa atividade bactericida e fagocitose. A opsonização também é deficiente e está relacionada a superexpressão do gene que codifica a superóxido dismutase1 (SOD-1) cujo lócus pertence ao cromossomo 21, levando a um aumento de ânions e reduzida eliminação de bactérias pela falta de radicais superóxido no meio celular.
 cujo lócus pertence ao cromossomo 21, levando a um aumento de ânions e reduzida eliminação de bactérias pela falta de radicais superóxido no meio celular.")
63
Desordens autoimunes Hipotireioidismo Doença celíaca? LES?
Alopécia areata Vitiligo Artropatia poli/pauciarticular com prevalência de 1%

64
Instabilidade e Subluxação Atlanto-Axial
A subluxação é uma importante complicação da síndrome que se caracteriza por fragilidade e alterações de ordem neurológica por compressão espinhal. Já a instabilidade atlanto-axial é assintomática e definida como um espaço maior que 4mm entre essas vértebras. Se devem provavelmente à elasticidade exagerada dos ligamentos. Tende a melhorar com a idade.

65
Doenças associadas à síndrome de Down

66
LEUCEMIA - Tanto a leucemia linfocítica aguda quanto a mielóide tem incidência aumentada na síndrome de Down, com um risco estimado de 10 a 20 vezes maior do que a população normal -Em particular, a leucemia megacarioblástica aguda ocorre com uma frequência 200 a 400 vezes maior - O aparecimento é bimodal (!), com o primeiro picos no recém nascido e novamente aos 3-6 anos de idade. O aumento de risco acompanha até a vida adulta.
, com o primeiro picos no recém nascido e novamente aos 3-6 anos de idade. O aumento de risco acompanha até a vida adulta.")
67
MECANISMOS PROPOSTOS 50% das crianças com síndrome de Down apresenta uma patologia mieloproliferativa transitória Há comprometimento da proliferação clônica local de células imaturas Ocorre resolução espontânea na maior parte dos casos, mas uma parcela evolui para leucemia aguda megacarioblástica, que é quase exclusiva da síndrome de Down essas alterações também são observadas em crianças fenotipicamente normais e mosaicismo com trissomia do 21 constitucional apesar da leucemia na síndrome de Down ser tipicamente descrita como megacarioblástica, também são encontrados colônias mielóides, eritróide e de macrófagos

68
MECANISMO Recém nascido com trissomia do 21 Doença mieloproliferativa
Comprometimento da proliferação clônica Resolução espontânea Aparecimento de leucemia megacarioblástica aguda

69
GENES ENVOLVIDOS AML/ PBP2 AML/TEL 1
- Faz parte da região crítica causadora da síndrome de Down - Envolvida em duas translocações cromossômicas associadas com leucemia: 1. 8; 21- AML (comum) 2. 3; 21- associada a algumas leucemias/mielodisplasias induzidas por quimioterapia AML/TEL 1 - Fusão gênica que resulta de t(12;21) é a aberração mais comum na leucemia linfoblástica aguda
 2. 3; 21- associada a algumas leucemias/mielodisplasias. induzidas por quimioterapia. AML/TEL 1. - Fusão gênica que resulta de t(12;21) é a aberração mais comum na leucemia linfoblástica aguda.")
70
GATA 1 - papel na maturação das células sanguíneas.
- 100% dos pacientes com síndrome de Down e com leucemia megacarioblástica aguda tiveram alteração no GATA1. Nenhum dos outros pacientes avaliados tiveram este gene anormal. -GATA1 é um fator de transcrição; ele controla a expressão de outros genes. -Regula genes que controlam a produção de células sanguíneas e plaquetas, que habilita o sangue a carregar o oxigênio e a coagular. -Estudos anteriores com camundongos mostraram que a perda do GATA1 fez com as células que dão origem às plaquetas a proliferarem excessivamente.

71
DOENÇA DE ALZHEIMER O cérebro de praticamente todos adultos com síndrome de Down e mais de 40 anos apresenta alterações fisiopatológicas caracterísitcas da doença de Alzheimer ( placas senis e massas neurofibrilares). A maior parte dos indíviduos não desenvolve um quadro clássico de Doença de Alzheimer, apresentando comumente um envelhecimento precoce e uma perda da capacidade de realizar as atividades habituais A prevalência da doença de Alzheimer é de 20 % para indivíduos normais na faixa etária de 75 a 85 anos, enquanto nos portadores da síndrome de Down ela ultrapassa os 75 % já na faixa dos 60 anos.
. A maior parte dos indíviduos não desenvolve um quadro clássico de Doença de Alzheimer, apresentando comumente um envelhecimento precoce e uma perda da capacidade de realizar as atividades habituais. A prevalência da doença de Alzheimer é de 20 % para indivíduos normais na faixa etária de 75 a 85 anos, enquanto nos portadores da síndrome de Down ela ultrapassa os 75 % já na faixa dos 60 anos.")
72
MECANISMOS PROPOSTOS Hetson et al (1977) – defeitos nos microtúbulos sugeridos por massas neurofibrilares - Harper et al (1979) - não conseguiram confirmar a existência de um defeito tubular sistêmico na doença de Alzheimer pela cultura de fibroblastos Nordenson et al (1980)- encontraram uma frequência aumentada de segmentos acêntricos no pacientes com doença de Alzheimer. Eles interpretaram isto como um achado consistente de uma proteína tubulina defetuosa levando a um mecanismo de “splindle” errático
 - não conseguiram confirmar a existência de um defeito tubular sistêmico na doença de Alzheimer pela cultura de fibroblastos. Nordenson et al (1980)- encontraram uma frequência aumentada de segmentos acêntricos no pacientes com doença de Alzheimer. Eles interpretaram isto como um achado consistente de uma proteína tubulina defetuosa levando a um mecanismo de splindle errático.")
73
CROMOSSOMOS ENVOLVIDOS NA DOENÇA DE ALZHEIMER
Cromossomo 14- gene da presenilina 1 ( PS1) Cromossomo 1- gene da presenilina 2 (PS2) Cromossomo 19 – gene da apf Cromossomo 21 – carreia o gene que codifica o precursor da B-amilóide . R elacionada a doença de início precoce, associação clara em alguns casos de doença de Alzheimer (região próxima ao centrômero) sem síndrome de Down. 3 produtos ligados ao Azlheimer: - proteína amilóide precursora - protéina beta derivada da glia - CuZn superóxido dismutase
 Cromossomo 1- gene da presenilina 2 (PS2) Cromossomo 19 – gene da apf. Cromossomo 21 – carreia o gene que codifica o precursor da B-amilóide . R elacionada a doença de início precoce, associação clara em alguns casos de doença de Alzheimer (região próxima ao centrômero) sem síndrome de Down. 3 produtos ligados ao Azlheimer: - proteína amilóide precursora. - protéina beta derivada da glia. - CuZn superóxido dismutase.")
74
MECANISMO PROPOSTO Cromossomo 21 extra
Neurotoxidade e morte dos neurônios Aumento da APP Depósito Aumento da beta amilóide Formação de placas senis

75
DOENÇA DE HIRSCHSPRUNG
Duas mal formações associadas a síndrome de Down: Atresia duodenal : 2 a 5 % com SD e 20 – 30% dos com AD tem SD Doença de Hirchsprung:2 com SD e 5-15% com DH tem SD Alguns pacientes apresentam as duas patologias Garver et al- Analisou 134 casos analisados, 103 tinham doença de segmento curto e 31 tinham aganglionose de longo segmento. Encontrou uma incidência de 5,8%. Quinn et al - encontraram uma incidência de 10-15%

76
Definição A doença de Hirschsprung ou megacólon aganglionar é uma doença congênita caracterizada pela ausência de gânglios entéricos em um segmento variável de intestino. Genética - Por um longo tempo foi considerado como uma doença multifatorial com uma possível relação com um gene autossômico recessivo - Com um aumento da sobrevida dos pacientes, famílias com herança dominante foram relatadas. Estudos de ligação ( linkage), detalhados posteriormente, embasaram a herança autossômica dominante dessa patologia.
, detalhados posteriormente, embasaram a herança autossômica dominante dessa patologia.")
77
GENES ENVOLVIDOS - A doença de Hirschsprung na Síndrome de Down é supostamente conseqüência de uma mutação no gene do GDNF (fator de crescimento derivado de células gliais). - Múltiplos loci parecem estar envolvidos, incluindo cromossomos 21q22 - Mudanças tanto no receptor tyrosine kinase (RET) proto-oncogene e endothelin-B receptor (EDNRB) gene foram evidenciadas em pacientes com trissomia do 21 - Há evidências genéticas que RET e EDNRB estão envolvidos na patogênese da doença de Hirschsprung
. - Múltiplos loci parecem estar envolvidos, incluindo cromossomos 21q22. - Mudanças tanto no receptor tyrosine kinase (RET) proto-oncogene e endothelin-B receptor (EDNRB) gene foram evidenciadas em pacientes com trissomia do Há evidências genéticas que RET e EDNRB estão envolvidos na patogênese da doença de Hirschsprung.")
78
RELAÇÃO COM OUTRAS CROMOSSOMOPATIAS
As trissomia dos cromossomos 21, 18 e 13 são as que mais comumente afetam nascidos com vida (menos de 1%) - Em abortos cariotipados representam 20% ( tipo mais comum de anormalidade cromossômica ) - Somente 25% dos zigotos com trissomia do 21 chegam a termo, sendo que este número cai para 5 % na trissomia do 18 e para 2,5% na trissomia do 13. - Desta forma , as aneuploidias são letais em seres humanos , sendo que os portadores de trissomia são uma pequena minoria de um grupo maior e mais heterogêneo.
 - Em abortos cariotipados representam 20% ( tipo mais comum de anormalidade cromossômica ) - Somente 25% dos zigotos com trissomia do 21 chegam a termo, sendo que este número cai para 5 % na trissomia do 18 e para 2,5% na trissomia do Desta forma , as aneuploidias são letais em seres humanos , sendo que os portadores de trissomia são uma pequena minoria de um grupo maior e mais heterogêneo.")
79
Triagem e Diagnóstico pré-natal da síndrome de Down

80
Por que realizar triagem?
Custos em relação aos exames diagnósticos Riscos de complicações Seleção de casos para realização de amniocentese OBS.: Não substituem exames diagnósticos

81
Marcadores plasmáticos utilizados
AFP: produzida no saco de Yolk e fígado fetal diminuída em 25% em gestações com Down (entre 15ª e 22ª semanas) μE3: hormônio produzido pela placenta diminuído em 25% no Down
 μE3: hormônio produzido pela placenta. diminuído em 25% no Down.")
82
Marcadores plasmáticos utilizados
HCG: hormônio produzido pela placenta níveis de β-HCG livre maiores que o dobro no Down(2,22 MoM) Inibina A: 1,79 MoM em torno da 16ª semana em gestações afetadas PAPP-A: menos da metade do normal em gestações afetadas (0,43 MoM) em torno da 16ª semana
 Inibina A: 1,79 MoM em torno da 16ª semana em gestações afetadas. PAPP-A: menos da metade do normal em gestações afetadas (0,43 MoM) em torno da 16ª semana.")
83
Efeito da idade materna
O aumento da idade materna aumenta as taxas de detecção e de falsos positivos obtidas pela triagem.

84
Efeito da idade materna

85
Efeito da ultrassonografia fetal
Maior precisão da idade gestacional, otimizando os resultados dos marcadores plasmáticos. Achados passíveis de associação com o Down: cistos de plexo coróide, intestino ecogênico, foco ecogênico intracardíaco, pielectasia. não são capazes de discriminar gestações comprometidas de gestações normais (Smith-Bindman, 2001).
.")
86
Efeito da ultrassonografia fetal
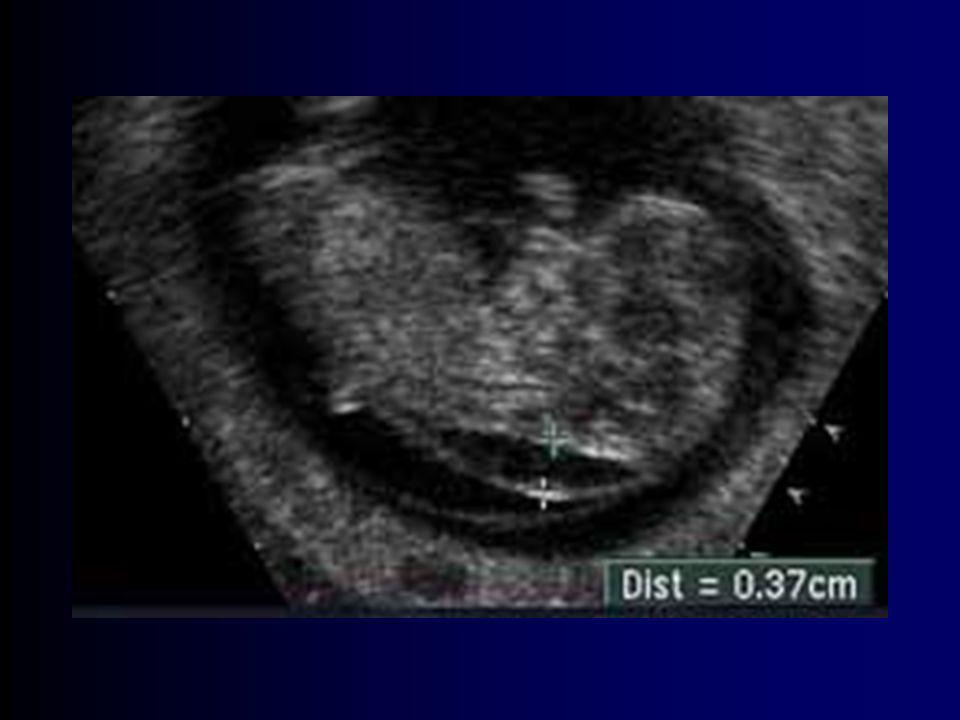
Transluscência nucal 15% tem síndrome de Down não faz diagnóstico (baixa sensibilidade) “Mesmo as melhores combinações de achados ultrassonográficos e outras variáveis são apenas preditivas, e nunca diagnósticas”.
 Mesmo as melhores combinações de achados ultrassonográficos e outras variáveis são apenas preditivas, e nunca diagnósticas .")
87
Outros fatores atuando sobre os marcadores
Peso materno Gestações com gêmeos Etnias Tabagismo

88
Triagem no 2º trimestre de gestação
Marcadores: AFP, μE3, HCG, idade materna (teste triplo); + inibina A (teste quádruplo) Período: 15ª - 22ª semanas Resultados: detecção do Down entre 60% e 80% falsos positivos: 3% a 7% (obs.: varia junto com detecção) abortamentos decorrentes da amniocentese: 0,9% 5% de mulheres submetidas à amniocentese
; + inibina A (teste quádruplo) Período: 15ª - 22ª semanas. Resultados: detecção do Down entre 60% e 80% falsos positivos: 3% a 7% (obs.: varia junto com detecção) abortamentos decorrentes da amniocentese: 0,9% 5% de mulheres submetidas à amniocentese.")
89
Triagem no 2º trimestre de gestação
Ponto de corte: 1:250 Maior (alto risco) Menor (baixo risco) Risco de gestação com síndrome de Down maior que risco da amniocentese Risco da amniocentese maior que risco de gestação downiana AMNIOCENTESE
 Menor (baixo risco) Risco de gestação com síndrome de Down maior que risco da amniocentese. Risco da amniocentese maior que risco de gestação downiana. AMNIOCENTESE.")
90
Triagem no 1º trimestre de gestação
Marcadores: PAPP-A, β-HCG livre, idade materna, transluscência nucal (teste combinado) Período: entre 8ª e 14ª semanas Resultados: taxa de detecção do Down: 81% a 88% taxa de falsos positivos: entre 3% e 7% amniocentese em 5% da população triada Detecção precoce × gestações que serão abortadas
 Período: entre 8ª e 14ª semanas. Resultados: taxa de detecção do Down: 81% a 88% taxa de falsos positivos: entre 3% e 7% amniocentese em 5% da população triada. Detecção precoce × gestações que serão abortadas.")
91
Triagem integrada Objetivos: Método: Resultados (ponto de corte 1:120)
reduzir número de falsos positivos, testes invasivos (amniocentese) e suas complicações (abortamentos). Método: associação dos resultados dos marcadores de 1º e 2º trimestres. Resultados (ponto de corte 1:120) detecção de 85% falsos positivos de 0,9% odds: integrado - 1:9 combinado - 1:45 quádruplo - 1:88 redução de amniocenteses em 4/5
 e suas complicações (abortamentos). Método: associação dos resultados dos marcadores de 1º e 2º trimestres. Resultados (ponto de corte 1:120) detecção de 85% falsos positivos de 0,9% odds: integrado - 1:9. combinado - 1:45. quádruplo - 1:88. redução de amniocenteses em 4/5.")
92
Comparação entre os métodos de triagem

93
Métodos de diagnóstico pré-natal

94
Amniocentese: Conceito: coleta de líquido amniótico por punção trans-abdominal guiada por ultrassonografia, submetendo-o a testes de análise cromossômica (numérica e estrutural).
.")
95
Amniocentese: Entre 14ª e 18ª semanas; Resultado em 2 semanas;
Efeitos adversos: abortamento: acréscimo em 0,5% a 1% cólicas, sangramentos, infecção e falta de líquido Ponto de corte :250 (no momento da triagem ou previsto para final da gestação??)
")
96
Amostragem de vilosidade coriônica
Amostras de placenta jovem por: punção trans-abdominal aspiração vaginal

97
Amostragem de vilosidade coriônica
entre 10ª e 12ª semanas mesmas complicações da amniocentese risco de abortamento: 3% a 5% amniocentese × amostra de placenta escolha cabe ao médico + paciente

98
Consideração final “ Por mais experimentado que seja o método de triagem pré-natal, seus resultados são sempre preditivos e nunca diagnósticos.”

99
CONDUTA

100
CUIDADOS ESPECIAIS Cardiopatia: DEVEM SER CORRIGIDAS CIRURGICAMENTE
40 a 50% têm cardiopatias congênitas Mais freqüentes: defeitos no septo ventricular ou atrial, tetralogia de Fallot e regurgitação aórtica. DEVEM SER CORRIGIDAS CIRURGICAMENTE

101
CUIDADOS ESPECIAIS Dificuldade de aprendizado
avaliar sua capacitação para o aprendizado, como: a acuidade auditiva acuidade visual. Não é raro um comprometimento nessas áreas. Na área oftalmológica há mesmo a possibilidade, maior que a usual, da existência de catarata ou glaucoma. Com grande freqüência, essas são crianças, que apresentam estrabismo e vícios de refração, como miopia e hipermetropia..

102
CUIDADOS ESPECIAIS Dieta: favorecer a mobilidade intestinal. oferecer uma dieta controlada e rica em fibras, com frutas e verduras. Já que uma dieta hipercalórica e desequilibrada somada à hipotonia favorecem à obesidade. Amamentação com o leite materno: se é importante para qualquer criança, para o portador da síndrome é fundamental. Isso porque melhora sua capacitação fono-deglutatória futura, melhora a mandíbula, favorece a deglutição e a sucção - o que leva mais tarde a uma melhor comunicação verbal, dado importantíssimo, uma vez que uma de suas características é a linguagem comprometida.

103
ASPECTOS IMPORTANTES Os portadores da Síndrome de Down idosos, como qualquer idoso, devem ter uma atenção a sua saúde. O aspecto psicológico dos pais também deve ser trabalhado.

105
Conclusão A expectativa de vida: 1900: 5 anos; 1950: 15 anos;
2000: similar a população em geral;

106
Os responsáveis pelo avanço:
Educadores; Profissionais de saúde; Associações; Persistência dos pais.

107
A discriminação Cultura fotográfica; Tratamento infantilizado;
Falta de crédito na habilitação social; Falta de crédito na habilitação laboral.

108
Casamento e Sexualidade
Convivência social Casamento e Sexualidade

109
A realidade A porcentagem de portadores de Síndrome de Down apresenta variações em alguns países; Doença de Alzheimer.

110
O alicerce do tratamento:
Capacitação Aprendizado Sociabilização Adequação Direitos

111
“Quanto tempo precisaremos para a sociabilização integral dos portadores da Síndrome de Down?”

Apresentações semelhantes














 Um processo especial.>")






