Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Hospital Materno Infantil de Brasília
Hospital Regional da Asa Sul Residência Médica em Terapia Intensiva Pediátrica Sessão de Anatomia Clínica: Síndrome de Imunodeficiência combinada severa (SCID) Apresentação:Luana Maia UTI Pediátrica HRAS/HMIB/SES/ DF Coordenação: Alexandre Serafim, Melissa Iole Da Cás Vita Coordenação Geral: Martha David Rocha de Moura Brasília, 4 de agosto de 2015
 Apresentação:Luana Maia. UTI Pediátrica HRAS/HMIB/SES/ DF. Coordenação: Alexandre Serafim, Melissa Iole Da Cás Vita. Coordenação Geral: Martha David Rocha de Moura. Brasília, 4 de agosto de")
2
TAPC, 1 ano e 1 mês, sexo feminino
Natural de Taguatinga- DF Residente em Brazlândia-DF Procedente do PSI HMIB Admissão na UTIP: 16/05/14 Motivo da internação: Insuficiência Respiratória Aguda (Síndrome do desconforto respiratório agudo-SDRA + pneumonia)
")
3
Resumo Clínico: Paciente internada desde 23/4/14 (HRBz), devido pneumonia e sibilância associada à desnutrição severa, diarréia crônica e monilíase oral/ perineal, além de linfopenia persistente. Recebeu ampicilina, gentamicina, hidrocortisona e fenoterol. Evoluiu com perda de peso progressiva e persistência do quadro diarréico. Foi transferida ao HRAS/HMIB no dia 6/5. Na admissão foi iniciado desmame da hidrocortisona, posteriormente substituída por prednisolona, substituído fenoterol por salbutamol e suspenso gentamicina. O esquema antibiótico foi substituído por ceftriaxona em 10/5 devido a piora radiológica e associado, em 12/5, azitromicina. Em 13/05, trocado ceftriaxona por cefepime e realizado concentrado de hemácias. Em 15/5, evoluiu com piora do estado geral , desidratação e desconforto respiratório, não responsiva à expansões volêmicas, salbutamol, concentrado de hemácias e oxigenoterapia. Permaneceu grave, foi intubada e acoplada à ventilação mecânica (VM) . Em 16/06 deu entrada na UTI Pediátrica.
 . Em 16/06 deu entrada na UTI Pediátrica.")
4
Admitida em uso de: - Dipirona SOS - Dieta zero - HV com 100% Holliday
- Midazolan 0,4 mg/kg/h - Fentanil 4 mcg/kg/min - Cefepime 150mg/kg/dia D3 - Azitromicina 10mg/kg/dia D4 - Bactrim 100 mg/kg/dia D2 - Fluconazol 8mg/kg/dia D2 - Prednisolona 10mg/dia (desmame) - Salbutamol 0,1mcg/kg/min - Omeprazol 1mg/kg/dia - Dipirona SOS
 - Salbutamol 0,1mcg/kg/min. - Omeprazol 1mg/kg/dia. - Dipirona SOS.")
5
História pregressa: Criança nascida de parto cesáreo com 39 semanas, com kg, 49cm, PC de 34cm e Apgar 9/10, teve alta com 2 dias de vida sem complicações. Pré-natal com 7 consultas com relato em cartão da criança de sorologias normais. Aleitamento exclusivo até os 6 meses quando introduziu leite integral com Mucilon, papa salgada e frutas. Mãe relata que até os 9 meses criança apresentava desenvolvimento normal, mas que há 3 meses criança passou a apresentar internações frequentes, perda de peso progressiva (4 kg em 3 meses) e diarréia. Vacinas atualizadas.
 e diarréia. Vacinas atualizadas..")
6
História social: Mora com mãe e pai em casa de alvenaria com 6 cômodos com saneamento básico. Sem animais de estimação. Nega viagens recentes. História familiar: Mãe 22 anos hígida, pai 27 anos com “anemia”, sem acompanhamento. Avós paternos e maternos com hipertensão arterial sistêmica e avó paterna com diabetes melitus. Nega câncer ou tuberculose na família.

7
Evolução 16/05 Hemodinamicamente estável sem drogas
Optado por manter antibioticoterapia com cefepime e fluconazol Recebeu expansão volêmica com cristalóide devido a ecografia funcional ter mostrado veia cava inferior colabada Apresentava quadro de SDRA grave Iniciada dieta enteral( formula de aminoácidos livres) Iniciada nistatina tópica em períneo
 Iniciada nistatina tópica em períneo.")
8
Evolução 17/05 Realizada troca do TOT(4,5 sem cuff por 4,5 com cuff) devido a escape com prejuízo à VM Otimizada VM e colhida secreção traqueal para cultura Apresentou sinais de hipovolemia e recebeu expansão volêmica com cristalóide

9
Evolução 18/05 Gasometria arterial com piora nos índices ventilatórios, mantendo quadro de SDRA grave Colocada em posição prona Otimizada VM para o quadro

10
Evolução 19/05 Ampliada antibioticoterapia (meropenen+vancomicina) – pancitopenia mantida com quadro grave Otimizada sedação – agitação com prejuízo à VM Gasometria arterial com quadro de SDRA mantido Coletado IgA anti TTG (pesquisa doença celíaca) Piora hemodinâmica (tempo de enchimento capilar de 4 seg + pulsos finos) - iniciada dobutamina (5mcg/kg/min) 10
 Piora hemodinâmica (tempo de enchimento capilar de 4 seg + pulsos finos) - iniciada dobutamina (5mcg/kg/min) 10.")
11
Evolução 20/05 Suspensa dobutamina (hemodinâmica estabilizada)
Edema mais importante – 1 dose de furosemida com boa resposta À noite com piora hemodinâmica (queda da PA, piora na perfusão e pulsos finos) – retornada dobutamina 5mcg/kg/min Recebeu nova dose de furosemida por diminuição na diurese com balanço hídrico positivo
 – retornada dobutamina 5mcg/kg/min. Recebeu nova dose de furosemida por diminuição na diurese com balanço hídrico positivo.")
12
Evolução 21/05 Iniciada furosemida intermitente (2mg/kg/dia)
Solicitada pesquisa de Pneumocistys jiroveci na secreção traqueal Tolerando redução lenta dos parâmetros ventilatórios Cursando com episódios mais numerosos de diarréia Piora ventilatória ao manuseio – retornada para posição prona

13
Evolução 22/05 Ecografia funcional mostrou veia cava colabada – optado por suspender furosemida e expansão com albumina Exames com manutenção de pancitopenia

14
Evolução 23/05 Secreção traqueal - Enterobacter cloacae sensível a meropenem - Entecoccus faecalis sensível a vancomicina VM com parâmetros altos e índices ventilatórios em piora Associado Sulfametoxazol-trimetoprim – leucopenia + falta de evidência de melhora clinica Metilprednisolona 2 mg/kg/d para tratamento da SARA Infusão de Albumina (alvo de albumina de 3,0) para melhor compensação hemodinâmica Iniciada GM-CSF (melhora leucocitária e pulmonar)
 para melhor compensação hemodinâmica. Iniciada GM-CSF (melhora leucocitária e pulmonar)")
15
Evolução 24/05 Iniciada nutrição parenteral parcial (manutenção de diarréia com redução da dieta enteral??) Retornada furosemida (edema) Recebeu nova expansão com albumina(albumina alvo = 3) Sem GM-CSF no Hospital Substituído fluconazol por micafungina Evoluindo estável hemodinamicamente com dobutamina(5),demandando ajustes ventilatórios devido a instabilidades
 Recebeu nova expansão com albumina(albumina alvo = 3) Sem GM-CSF no Hospital. Substituído fluconazol por micafungina. Evoluindo estável hemodinamicamente com dobutamina(5),demandando ajustes ventilatórios devido a instabilidades.")
16
Evolução 25 e 26/05 Solicitado parecer à Imunologia
Mantendo quadro de diarréia volumosa com numerosos episódios Monilíase perineal com melhora Pesquisa de P. jiroveci – negativa Dieta modificada para fórmula hidrolisada após discussão com gastroped Trocado cateter venoso central(VSCD – VJIE)- enviada ponta para cultura Exames com pancitopenia mantida – concentrado de hemácias Correção de Magnésio( nível de 1,5 mEq/l) Apresentou fezes escurecidas
- enviada ponta para cultura. Exames com pancitopenia mantida – concentrado de hemácias. Correção de Magnésio( nível de 1,5 mEq/l) Apresentou fezes escurecidas.")
17
Evolução 27 e 28/05 Ecoscopia da veia cava inferior mostrou colabamento – expansão com cristalóide Iniciada butrição parenteral total plena com dieta zero Associado dexmedetomedina(0,3mcg/kg/h) à sedação para otimização
 à sedação para otimização.")
18
Evolução 29/05 Discutido com gastroped que orientou iniciar IBP com nutrição parenteral total plena e dieta zero por 72h Infectologia orientou iniciar ciprofloxacino A tarde cursou com piora respiratória necessitando pushs de curare e otimização da sedoanalgesia, com boa resposta Substituída dobutamina por milrinona (0,5mcg/kg/min)
")
19
Evolução 30/05 Melhora da diarréia e dermatite perineal após suspensão da dieta Evoluindo com melhora hemodinamica Melhora progressiva no leucograma (manutenção de linfopenia) Necessitou correção de Potássio(2,5) Realizada TC de tórax
 Necessitou correção de Potássio(2,5) Realizada TC de tórax.")
20
Evolução 31/05 Retornada furosemida intermitente (piora no edema com rebaixamento do fígado com BH positivo) Mantendo saturações mais baixas(75-85%), lábil às manipulações (dessaturações ate 60%) Recebeu reposição de Magnésio(1,5) Piora na anemia(Hb7,5) e plaquetopenia (26.000) – recebeu concentrado de hemácias com discreta melhora hemodinâmica
, lábil às manipulações (dessaturações ate 60%) Recebeu reposição de Magnésio(1,5) Piora na anemia(Hb7,5) e plaquetopenia (26.000) – recebeu concentrado de hemácias com discreta melhora hemodinâmica.")
21
Evolução 01/06 Pico hipertensivo + piora da perfusão + queda da FC(88) + dessaturação(70%) – aumentada milrinona (0,5- 0,75) e associada epinefrina (0,1mcg/kg/min) Cursou com novo pico pressórico sendo suspensa epinferina Apresentou melena Exames com piora na plaquetopenia(14.000) com hipocalemia – recebeu concentrado de plaquetas + correção de potássio Apresentou enterorragia, com piora hemodinâmica – recebeu plasma + concentrado de hemácias Sedação novamente otimizada por agitação (fentanil 4mcg/kg/h + midazolan 0,6mg/kg/h + ketamina 20 mcg/kg/min)
 + dessaturação(70%) – aumentada milrinona (0,5- 0,75) e associada epinefrina (0,1mcg/kg/min) Cursou com novo pico pressórico sendo suspensa epinferina. Apresentou melena. Exames com piora na plaquetopenia(14.000) com hipocalemia – recebeu concentrado de plaquetas + correção de potássio. Apresentou enterorragia, com piora hemodinâmica – recebeu plasma + concentrado de hemácias. Sedação novamente otimizada por agitação (fentanil 4mcg/kg/h + midazolan 0,6mg/kg/h + ketamina 20 mcg/kg/min)")
22
Evolução 02/06 Novo episódio de enterorragia com resíduo gástrico escurecido – novo concentrado de plaquetas Muito secretiva demandando muitas aspirações Recebeu novo concentrado de hemácias (palidez, taquicardia,enterorragia) Iniciado octreotide (1mcg/kg/h- diminuir diarréia e sangramento gastrointestinal) Discutido caso com hemato e imunoped – iniciado Bactrin profilático e Imunoglobulina humana após coleta de Imunogobulinas
 Iniciado octreotide (1mcg/kg/h- diminuir diarréia e sangramento gastrointestinal) Discutido caso com hemato e imunoped – iniciado Bactrin profilático e Imunoglobulina humana após coleta de Imunogobulinas.")
23
Evolução 02/06 Realizada punção de artéria femoral D para monitorização de PAi – recebeu novo concentrado de plaquetas antes (mantinha evidencias de sangramento com resíduo gástrico escurecido) Mantendo sangramento gastrointestinal a noite com queda de Hb – novo concentrado de hemácias IgM,IgG e IgE <P3 com IgA normal Manteve linfopenia durante toda a internação
 Mantendo sangramento gastrointestinal a noite com queda de Hb – novo concentrado de hemácias. IgM,IgG e IgE <P3 com IgA normal. Manteve linfopenia durante toda a internação.")
24
Evolução 03/06 Gasometria arterial demonstrava manutenção de SDRA grave Resultado TC tórax (30/05): Opacidade em vidro fosco difusa e bilateral com espessamento liso de septos inter e intralobulares, além de esparsas opacidades alveolares. Não há sinais de lesões nodulares ou cavitárias. A distribuição broncovascular tem aspecto anatômico em ambos os pulmões. Ausência de derrame ou espessamento pleural. Mediastino com aspecto anatômico, sem evidência de linfonodomegalias. Estruturas ósseas incluídas de aspecto normal.
: Opacidade em vidro fosco difusa e bilateral com espessamento liso de septos inter e intralobulares, além de esparsas opacidades alveolares. Não há sinais de lesões nodulares ou cavitárias. A distribuição broncovascular tem aspecto anatômico em ambos os pulmões. Ausência de derrame ou espessamento pleural. Mediastino com aspecto anatômico, sem evidência de linfonodomegalias. Estruturas ósseas incluídas de aspecto normal.")
25
Evolução 04/06 Colhida secreção traqueal para cultura
IgA anti-TTG: 0,63U (VR<20U) Palpada massa em fossa ilíaca D – solicitada ecografia abdominal : Fígado com dimensões aumentadas e ecotextura levemente heterogênea. Não há sinais de dilatação das vias biliares . Vesícula biliar distendida, sem evidências de cálculos. Rins, baço e pâncreas anatômicos. Área hipoecóica irregular, mal definida, na fossa ilíaca direita , medindo cerca de 3,4x3,3cm, que pode representar coleção ou alça intestinal aperistáltica distendida. Impressão de espessamento parietal de alças colônicas, sobretudo do reto.
 Palpada massa em fossa ilíaca D – solicitada ecografia abdominal : Fígado com dimensões aumentadas e ecotextura levemente heterogênea. Não há sinais de dilatação das vias biliares . Vesícula biliar distendida, sem evidências de cálculos. Rins, baço e pâncreas anatômicos. Área hipoecóica irregular, mal definida, na fossa ilíaca direita , medindo cerca de 3,4x3,3cm, que pode representar coleção ou alça intestinal aperistáltica distendida. Impressão de espessamento parietal de alças colônicas, sobretudo do reto.")
26
Evolução 05/06 Realizada coleta de amostra para CD4 e CD8
1 episódio de melena Piora ventilatória mesmo com altos parâmetros (dP de 13 ; PEEP de 14 ; FiO2 de 85% FR de 25) necessitando aumento de FiO2 para 90% Cultura de Secreção Traqueal(03/06): K. pneumoniae, sensível apenas à Amicacina e à Colistina.
 necessitando aumento de FiO2 para 90% Cultura de Secreção Traqueal(03/06): K. pneumoniae, sensível apenas à Amicacina e à Colistina.")
27
Evolução 06/06 Suspenso ciprofloxacino e iniciada amicacina
Coletada amostra para pesquisa de BAAR no lavado gástrico (negativa 2 amostras) Reiniciada dieta enteral Apresentou enfisema subcutâneo em pescoço e 1/3 superior de tórax Rx de tórax mostra áreas de enfisema mediastinal em terço superior do tórax mais acentuado à direita. Existe área de hipertransparência em base pulmonar direita, mas com presença de parênquima pulmonar. Opacidades alveolo-intersticiais bilaterais. Conduta expectante (pela CIPE)
 Reiniciada dieta enteral. Apresentou enfisema subcutâneo em pescoço e 1/3 superior de tórax. Rx de tórax mostra áreas de enfisema mediastinal em terço superior do tórax mais acentuado à direita. Existe área de hipertransparência em base pulmonar direita, mas com presença de parênquima pulmonar. Opacidades alveolo-intersticiais bilaterais. Conduta expectante (pela CIPE)")
28
Evolução 07-08 e 09/06 Recebeu concentrado de hemácias
Piora ventilatória demandando aumento de parâmetros(dP 23 ; PEEP 15 ; FiO2 100%) Colhida amostra para contagem de CD3, CD19, CD56 Suspensa cetamina continua PPD(05/06): Não reator Suspensos octreotide,vancomicina(D22) e clorpromazina Timo involuído ao Raio X
 Colhida amostra para contagem de CD3, CD19, CD56. Suspensa cetamina continua. PPD(05/06): Não reator. Suspensos octreotide,vancomicina(D22) e clorpromazina. Timo involuído ao Raio X.")
29
Evolução 09/06 Avaliada pela imunologia que orientou:
Colher dosagens de CD19 e CD56. CD4 e CD8 já colhidos. Dosar IgG novamente após 15 dias da infusão de imunoglobulina. Caso IgG < 600, fazer nova infusão de imunoglobulina. Se necessário novas transfusões optar por hemoderivados irradiados Manter Sulfametoxazol- Trimetropin contínuo Deixar criança em isolamento Se possível, tentar realizar coprocultura SD: Imunodeficiência a esclarecer ( linfopenia e Igs baixas)
")
30
Evolução 10/06 Adiada broncoscopia devido as condições clínicas (VM com altos parâmetros) Aumento do enfisema subcutâneo com acometimento de região periorbitária E e Raio X mostrando aumento do pneumomediastino – Síndrome de escape de ar

31
Evolução 11/06 Durante a madrugada com hipoxemia grave e refratária além de piora no enfisema, com descompensação hemodinâmica PCR sem resposta após 5 doses de adrenalina Óbito constatado as 4:40h. Mãe esteve à beira do leito durante todo o período. Exames: Células NK (CD 56): 277/mm3 (VR: /mm3);P 10-50 Linfócitos B (CD 19): 27/mm3 (VR: /mm3); P<10 Linfócitos T (CD3): 319/mm3 (VR: /mm3); P<10
: 277/mm3 (VR: /mm3);P Linfócitos B (CD 19): 27/mm3 (VR: /mm3); P<10. Linfócitos T (CD3): 319/mm3 (VR: /mm3); P<10.")
32
Dra. Melissa Iole Da Cás Vita Núcleo de Anatomia Patológica
Sessão de Anatomia Clínica Hospital Materno Infantil de Brasília Dra. Melissa Iole Da Cás Vita Núcleo de Anatomia Patológica

33
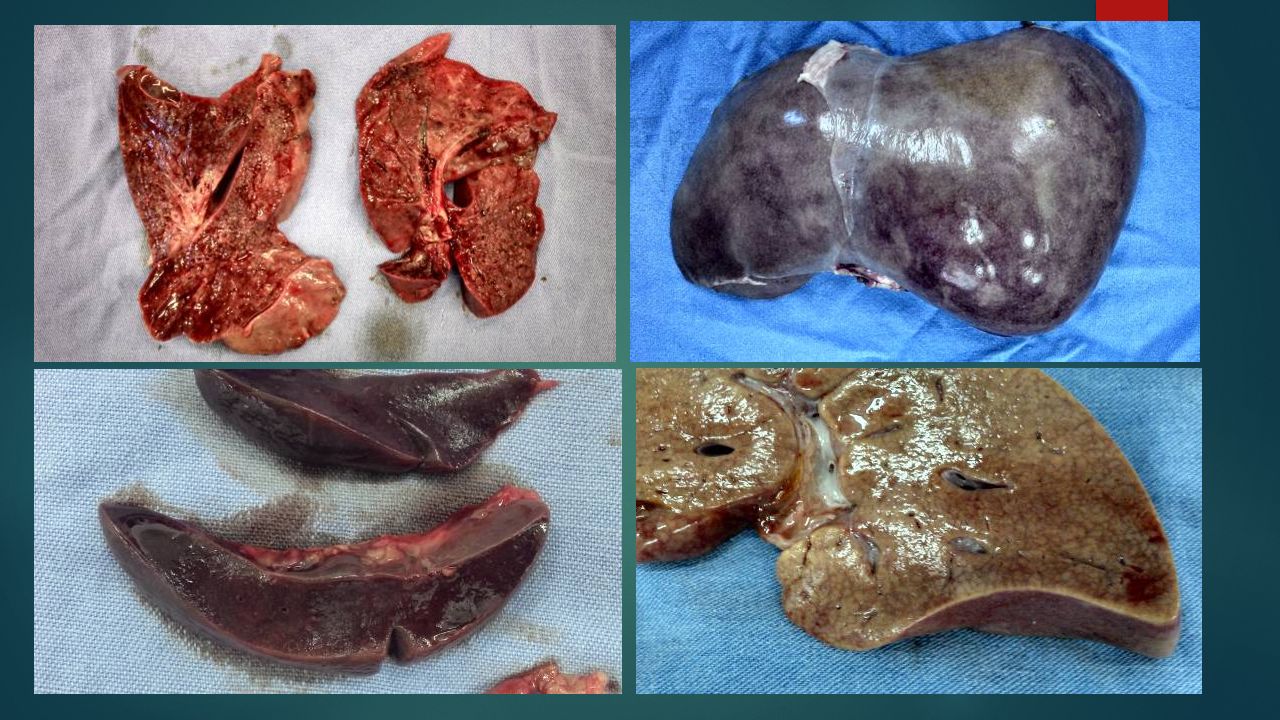
A : exame externo T.A.P.C., cadáver do sexo feminino, com desnutrição grave e história de infecções recorrentes e leucopenia persistente Edema generalizado, redução do panículo adiposo. Lesões eritemato-descamativas em região perineal Lesões eritematosas e fissuras “sangrantes” em borda anal Punções periféricas em regiões cubital e femoral

34
Distensão de alças intestinais
Hepatomegalia Área cardíaca aumentada Ausência de timo

35
A.044.14: Exame interno Derrame seroso pleural e pericárdico
Timo: ausência de tecido tímico (depleção) Pulmões (PD: 160g e PE: 142g): aspecto miliar discreto. Consistência borrachosa e sólida, predominantemente á direita Coração (48g): área cardíaca aumentada, dilatação do VD Traquéia: mucosa avermelhada e irregular Fígado: pardo-amarelado, aumentado de volume (6cm do RCD e 3,5cm do AX)
 Pulmões (PD: 160g e PE: 142g): aspecto miliar discreto. Consistência borrachosa e sólida, predominantemente á direita. Coração (48g): área cardíaca aumentada, dilatação do VD. Traquéia: mucosa avermelhada e irregular. Fígado: pardo-amarelado, aumentado de volume (6cm do RCD e 3,5cm do AX)")
37
A : exame interno Rins (Rim D + Rim E: 92g): parênquima pálido e com limite córtico-medular evidente Baço (15g); friável, difluente e vinhoso Estômago: enantema difuso do corpo Cólon: dilatado, com mucosa irregular, friável e avermelhada. Presença de deposição irregular de membrana pardo-amarelada e destacável na superfície mucosa. Deposição difusa de material fibrino-purulento no peritôneo Cérebro (700g): giros reduzidos de volume, congestão e edema
; friável, difluente e vinhoso. Estômago: enantema difuso do corpo. Cólon: dilatado, com mucosa irregular, friável e avermelhada. Presença de deposição irregular de membrana pardo-amarelada e destacável na superfície mucosa. Deposição difusa de material fibrino-purulento no peritôneo. Cérebro (700g): giros reduzidos de volume, congestão e edema.")
38
Ascite Colite (cólon dilatado, friável, com fissuras/úlceras e pseudomembrana Peritonite

39
A.044.14: microscopia Pulmão SDRA (“dano contínuo”): Fase exsudativa e
Proliferativa concomitantes Fibrose, metaplasia escamosa, penumócitos reativos Denso infiltrado inflamatório mono e polimorfonuclear brônquico, bronquiolar e alveolar

40
Pulmão Congestão, focos de hemorragia

41
Células gigantes intersticiais: infecção viral?
VSR, sarampo, parainfluenza vírus e VZV Pulmão Broncopneumonia: denso infiltrado inflamatório mono e polimorfonuclear brônquico, bronquiolar e alveolar

42
SDRA (“dano contínuo”): Fase exsudativa e Proliferativa concomitantes Membranas hialinas Dano alveolar difuso com formação de MH, fibrose intersticial, dano endotelial e micrombos vasculares

43
FCólon Colite aguda, com ulcerações da mucosa e formação de pseudomembrana inflamatória Ausência de tecido linfoide associada à mucosa e plasmócitos intraepiteliais

44
Peritonite aguda difusa: processo inflamatório agudo e
material fibrino-necrótico-leucocitário de serosa de bexiga, ovário, tubas uterinas, intestinos delgado e grosso. Ovário Cólon Infiltrado inflamatório em mesentério

45
Fígado: esteatose micro e macrogoticular
Linfonodos: depleção linfocítica, ausência de centros germinativos, degeneração lipoídica Fígado: esteatose micro e macrogoticular Rim: necrose tubular aguda

46
A.044.14 Síndrome de Imunodeficiência combinada severa (SCID)
Infecções plurimicrobianas Broncopneumonia Pneumonia de aspecto viral Colite, com pseudomembrana Peritonite fibrino-leucocitária Eczema fúngico em região perianal Involução tímica Depleção linfocitária em linfonodos e baço Ausência de centros germinativos nodais Ausência de MALT Hipoplasia esplênica SDRA Diarréia crônica Desnutrição grave Hipoproteinemia Edema de membros Cardiomegalia, com dilatação de VD Derrame pericárdico Derrame pleural Choque séptico Necrose tubular aguda Insuficiência cardíaca (?) Óbito inevitável
 Óbito inevitável.")
47
SCID Hospital Materno Infantil de Brasília
Hospital Regional da Asa Sul Residência Médica em Terapia Intensiva Pediátrica Luana Maia – R3 UTIP

48
SCID Constituem um grupo de imunodeficiência primária (IDP) que compartilham o fenótipo clínico de profunda deficiência nas funções imune celular e humoral. Principal característica: ausência completa ou número muito baixo de células T maduras. Outras características dependem da presença ou ausência de linfócitos B(T-B-/B+) e células natural killer(T-,NK-/NK+), padrão de herança, e o defeito genético-molecular.
 que compartilham o fenótipo clínico de profunda deficiência nas funções imune celular e humoral. Principal característica: ausência completa ou número muito baixo de células T maduras. Outras características dependem da presença ou ausência de linfócitos B(T-B-/B+) e células natural killer(T-,NK-/NK+), padrão de herança, e o defeito genético-molecular.")
49
SCID A prevalência de todos os tipos de SCID está calculada em cerca de 1/ a 1/ nascimentos São heranças genéticas (de forma autossômica recessiva ou de forma recessiva ligada ao cromossomo X) Afeta mais homens que mulheres Conduzem a uma susceptibilidade extrema a infecções graves Emergência Pediátrica
 Afeta mais homens que mulheres. Conduzem a uma susceptibilidade extrema a infecções graves Emergência Pediátrica.")
50
SCID Defeito na Cadeia Gama comum para IL-2, -4, -7, -9, -15, -21 (T-,B+,NK-) Deficiência na Adenosina Deaminase-ADA (T-,B-,NK-) Deficiência na cadeia alfa do receptor de IL-7 – proliferação de T CD4 (T-,B+,NK+) Deficiência na JAK-3 quinase – funcionamento cadeia Gama (T-,B+,NK-) Deficiências nas cadeias CD3-receptor Ag (T-,B+,NK+) Deficiência em CD45 (T-,B+,NK+) Deficiência em RAG-1 e 2 – receptor Ag ( T-,B-) Disgenesia Reticular (defeito AK2) – maturação (T-,B-) Deficiência na Artemis – recombinação e reparo no DNA (T-,B-)
 Deficiência na JAK-3 quinase – funcionamento cadeia Gama (T-,B+,NK-) Deficiências nas cadeias CD3-receptor Ag (T-,B+,NK+) Deficiência em CD45 (T-,B+,NK+) Deficiência em RAG-1 e 2 – receptor Ag ( T-,B-) Disgenesia Reticular (defeito AK2) – maturação (T-,B-) Deficiência na Artemis – recombinação e reparo no DNA (T-,B-)")
51
SCID- Apresentação clínica
Infecções persistentes e recorrentes por microrganismos oportunistas Monilíase oral ou perineal recorrente ou persistente sem ou com pouca resposta ao tratamento Tosse persistente Diarréia persistente com déficit de ganho pondero - estatural e desnutrição Doença enxerto x hospedeiro (Linfócitos T maternos ou hemocomponentes)
")
52
SCID - Diagnóstico Linfopenia (<2500/mm³) – deve ser confirmada
Contagem diferencial de linfócitos - CD3, CD4, CD8, CD19 e CD16/56 Função linfocitária Dosagem de imunoglobulinas séricas (IgG, IgM, IgA, IgE) Excluir HIV História familiar
 Excluir HIV. História familiar.")
53
SCID-Tratamento Isolamento para prevenir infecções oportunistas;
Não oferecer vacinas de vírus ou bactérias vivas ou atenuadas; Irradiação de todos os hemoderivados, para prevenir DECH; Todos os produtos hemoderivados devem ser de doadores CMV- negativos; Identificar e tratar todas infecções: usar culturas diretas ou técnicas PCR;

54
SCID - Tratamento Terapia de substituição com imunoglobulina (IGIV - Imunoglobulina Intravenosa) Transplante de Medula Óssea (antes de 3 meses com taxa de sucesso>95%) Terapia genética (diagnóstico molecular)
 Terapia genética (diagnóstico molecular)")
55
SCID- Perspectivas Triagem Neonatal para SCID
Debate nos EUA desde 2001 2008 o estado americano de Wisconsin foi pioneiro na triagem neonatal de SCID 2010 Secretaria de Saúde dos EUA aprovou a adição de SCID ao teste de triagem neonatal Triagem para SCID – quantificação de círculos excisados de células T (TRECs) – marcadores do desenvolvimento normal destas células Instituto de Ciências Biomédicas da USP e Escola Paulista de Medicina da UNIFESP Triagem neonatal
 – marcadores do desenvolvimento normal destas células. Instituto de Ciências Biomédicas da USP e Escola Paulista de Medicina da UNIFESP Triagem neonatal.")
56
Deficiências Humorais
Apresentação clínica: Infecções graves e/ou de repetição por bactérias encapsuladas; Infecções de repetição das vias aéreas Suspeitas Diagnósticas: Deficiências de Ac: agamaglobulinemia, imunodeficiência comum variável, deficiência de IgA, deficiência de anticorpos Diagnóstico diferencial: Deficiência de complemento Leucopenias e Doença granulomatosa Crônica Síndrome de Wiskott-Aldrich (plaquetopenia) Investigação: Hemograma: atenção ao número de linfócitos e neutrófilos Dosagem de Igs (IgG,IgA,IgM)
 Investigação: Hemograma: atenção ao número de linfócitos e neutrófilos. Dosagem de Igs (IgG,IgA,IgM)")
57
Deficiências Celulares
Apresentação clínica: Crescimento e desenvolvimento inadequados já presentes no 1° ano de vida Infecções graves/repetição por quaisquer germes inclusive oportunistas Suspeitas Diagnósticas: Imunodeficiência Combinada Grave Deficiência de Linfócitos T Síndrome de Wiskott-Aldrich Deficiência de CD40L Investigação: Hemograma- atenção ao número de linfócitos e neutrófilos Dosagem de Igs( IgG,IgA,IgM) Subpopulação de linfócitos: CD3, CD4, CD8, CD19 e CD16/56 Teste HIV, PPD e candidina
 Subpopulação de linfócitos: CD3, CD4, CD8, CD19 e CD16/56. Teste HIV, PPD e candidina.")
58
Defeitos de fagócitos Apresentação Clínica Suspeitas Diagnósticas:
Infecções piogênicas de repetição (principalmente Staphylococcus aureus) Infecções por Candida ou outro fungo Abscesso pulmonar Suspeitas Diagnósticas: Neutropenia Doença Granulomatosa Crônica Hiper IgE Investigação: Hemograma- atenção ao número de linfócitos e neutrófilos Dosagem de Igs( IgG,IgA,IgM,IgE) Teste de dihidrorodamina (DHR)
 Infecções por Candida ou outro fungo. Abscesso pulmonar. Suspeitas Diagnósticas: Neutropenia. Doença Granulomatosa Crônica. Hiper IgE. Investigação: Hemograma- atenção ao número de linfócitos e neutrófilos. Dosagem de Igs( IgG,IgA,IgM,IgE) Teste de dihidrorodamina (DHR)")
59
Referências Bibliográficas
Teixeira C et al. – Imunodeficiência combinada grave, Acta Pediatr Port 2011:42(2): (acesso em 18/06/15) Al-Herz et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency, Frontiers in Immunology. Primary Immunodeficiencies, Volume 5, Article 162,2014. Shearer et al. Establishing diagnostic criteria for SCID, leaky SCID and Omen Syndrome: The Primary Immune deficiency Treatment Consortium experience; J Allergy Clin Immunol,Volume 133,n.4,2014 IPOPI – International Patient Organization for Primary Immunodeficiencies; pdf (Acesso em 18/06/15) Kanegae MP et al. Triagem Neonatal para SCID; Rev. bras. alerg. imunopatol. Vol. 34. N° 1, 2011
: (acesso em 18/06/15) Al-Herz et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency, Frontiers in Immunology. Primary Immunodeficiencies, Volume 5, Article 162,2014. Shearer et al. Establishing diagnostic criteria for SCID, leaky SCID and Omen Syndrome: The Primary Immune deficiency Treatment Consortium experience; J Allergy Clin Immunol,Volume 133,n.4,2014. IPOPI – International Patient Organization for Primary Immunodeficiencies; pdf (Acesso em 18/06/15) Kanegae MP et al. Triagem Neonatal para SCID; Rev. bras. alerg. imunopatol. Vol. 34. N° 1,")
60
Nota do Editor do site, Dr. Paulo R
Nota do Editor do site, Dr. Paulo R. Margotto Consultem também Aqui e Agora! 2008 Monografia-2008 (Apresentação):SCID*-passado,presente e futuro (relato de caso de um paciente acompanhado no Hospital Regional da Asa Sul) Autor(es): Ana Carolina Monteiro Cunha OBJETIVO DESTA MONOGRAFIA Relatar um caso de SCID (1 ano e 4 meses, sexo masculino), acompanhado no Hospital Regional da Asa Sul pela equipe de imunologia e infectologia, e apresentar uma revisão sobre o tema, enfocando a importância do diagnóstico precoce e perspectivas futuras em relação ao tratamento. A fisiopatologia e biologia molecular podem variar, no entanto, a deficiência de linfócitos T e B é o ponto comum em todas as formas de SCID. São as características celulares que ajudam a diferenciar as várias formas de SCID. *SCID: se lê “squid”
:SCID*-passado,presente e futuro (relato de caso de um paciente acompanhado no Hospital Regional da Asa Sul) Autor(es): Ana Carolina Monteiro Cunha. OBJETIVO DESTA MONOGRAFIA. Relatar um caso de SCID (1 ano e 4 meses, sexo masculino), acompanhado no Hospital Regional da Asa Sul pela equipe de imunologia e infectologia, e apresentar uma revisão sobre o tema, enfocando a importância do diagnóstico precoce e perspectivas futuras em relação ao tratamento. A fisiopatologia e biologia molecular podem variar, no entanto, a deficiência de linfócitos T e B é o ponto comum em todas as formas de SCID. São as características celulares que ajudam a diferenciar as várias formas de SCID. *SCID: se lê squid")
61
Finalizando, é de extrema importância que o pediatra esteja muito atento aos principais pontos de alerta para se pensar em imunodeficiêncisa primária, de tal forma que os casos suspeitos sejam encaminhados para serviços de referência em imunologia o mais precocemente possível, possibilitando a adequada investigação diagnóstica e a adoção de medidas terapêuticas específicas, melhorando o prognóstico e a qualidade de vida destes pacientes. Além de acompanhamento para prevenção de possíveis complicações inerentes à doença genética. A síndrome da imunodeficiência combinada grave é, geralmente, considerada como sendo a mais grave das imunodeficiências primárias. Sem um transplante de medula óssea bem-sucedido ou a terapia genética, o doente está sujeito ao risco constante de uma infecção grave ou fatal. Com um transplante de medula óssea bem-sucedido, o sistema imunitário defeituoso do doente é substituído por um sistema imunitário normal e o funcionamento dos linfócitos T é restaurado. O primeiro transplante de óssea para SCID foi realizado em Esse doente está hoje vivo e bem de saúde.

62
(caso clínico neonatal)
2007 Caso Clínico: Imunodeficiência combinada grave (SCID) Autor(es): Danilo Hellou, Paulo R. Margotto, Fabíola Scancetti Tavares (caso clínico neonatal) História Clínica Primeiro filho do casal falecido aos seis meses de idade por pneumonia e septicemia (sic). Segundo filho do casal nasceu bem.Aos dois meses, após vacinação contra BCG apresentou supuração no local além de pneumonia e monilíase oral e perineal. Recebeu alta hospitalar com esquema tríplice e voltou após dois meses com a mesma pneumonia.Nesta ocasião foi revelado leucopenia severa e hipogamaglobulinemia quando foi diagnosticado Imunodeficiência combinada severa (SCID: se lê “squid”). Nasceu com 38 sem + 2 dias. Parto ocorrido no no automóvel. Peso 3320g PC: 34cm Est: 50cm Apgar de 8 e 10 em 1° e 5° min. Amamentando ao seio materno
 Autor(es): Danilo Hellou, Paulo R. Margotto, Fabíola Scancetti Tavares. (caso clínico neonatal) História Clínica. Primeiro filho do casal falecido aos seis meses de idade por pneumonia e septicemia (sic). Segundo filho do casal nasceu bem.Aos dois meses, após vacinação contra BCG apresentou supuração no local além de pneumonia e monilíase oral e perineal. Recebeu alta hospitalar com esquema tríplice e voltou após dois meses com a mesma pneumonia.Nesta ocasião foi revelado leucopenia severa e hipogamaglobulinemia quando foi diagnosticado Imunodeficiência combinada severa (SCID: se lê squid ). Nasceu com 38 sem + 2 dias. Parto ocorrido no no automóvel. Peso 3320g. PC: 34cm Est: 50cm. Apgar de 8 e 10 em 1° e 5° min. Amamentando ao seio materno.")
63
Antecedentes familiares
Pai e mãe consangüíneos: primos de 1° grau. Irmão: falecido aos 6 meses com pneumonia e septicemia(sic). Irmão: falecido aos 8 meses por pneumonia. Portador de SCID. Com os dados da história clínica, RN permaneceu em isolamento de contato Enviado amostra de sangue para Imunologia Pediátrica UNIFESP Baixíssimo n° de linfócitos. Pela história e exames complementares. Diagnosticado Imunodeficiência Combinada Severa(SCID) Conduta Antibioticoterapia/ imunoglobulina 21/21 dias/Ambulatório do Hospital de Base de Brasília
. Irmão: falecido aos 8 meses por pneumonia. Portador de SCID. Com os dados da história clínica, RN permaneceu em isolamento de contato. Enviado amostra de sangue para Imunologia Pediátrica UNIFESP Baixíssimo n° de linfócitos. Pela história e exames complementares. Diagnosticado Imunodeficiência Combinada Severa(SCID) Conduta. Antibioticoterapia/ imunoglobulina 21/21 dias/Ambulatório do Hospital de Base de Brasília.")
64
Sem as células T a vida não pode ser mantida;
2008 Imunodeficiência Combinada Grave Fabrício Prado Monteiro Sem as células T a vida não pode ser mantida; Uma ausência das células T funcionais causa a imunodeficiência combinada severa (grave); Grave porque é fatal e combinada porque, no homem, as células B não podem exercer sua função sem o auxílio das células T, de modo que mesmo que as células B não sejam diretamente afetadas pelo defeito, tanto a imunidade humoral quanto a celular são perdidas; A SCID é um fenótipo que pode resultar de qualquer um dentre vários defeitos genéticos diferentes e afeta mais homens que mulheres na proporção de 3:1
; Grave porque é fatal e combinada porque, no homem, as células B não podem exercer sua função sem o auxílio das células T, de modo que mesmo que as células B não sejam diretamente afetadas pelo defeito, tanto a imunidade humoral quanto a celular são perdidas; A SCID é um fenótipo que pode resultar de qualquer um dentre vários defeitos genéticos diferentes e afeta mais homens que mulheres na proporção de 3:1.")
65
Como a maioria das imunodeficiências primárias (ID) é transmitida por herança recessiva ligada ao X ou autossômica recessiva, as crianças tornam-se o grupo alvo destas patologias e o neonatologista tem um importante papel em reconhecer precocemente os sinais e sintomas dessas doenças para prover a melhor abordagem terapêutica, antes que as infecções graves ou dano tecidual permanente possam ocorrer. Estima-se que a incidência das ID primárias ocorre na proporção de 1:2000 nascidos vivos, embora possa ser mais elevada devido a muitas crianças morrerem de infecção antes da suspeição diagnóstica. A primeira doença de ID descrita foi reportada por Bruton há mais de 50 anos e, desde então, mais de 150 ID primárias têm sido descritas.

66
2013 Quando suspeitar de imunodeficiência primária no recém-nascido e lactente Autor(es): Marcia Cristina Mondaini Salazar, Fabíola Scancetti Tavares (Capítulo do livro Assistência ao Recém-Nascido de Risco, ESCS, Brasília, 3ª Edição, 2013) A história clínica de uma criança com suspeita de imunodeficiência (ID) deve abranger dados como doença materna, duração da gestação, peso e condições do nascimento, relato da queda do coto umbilical (> 21 dias), a natureza e a gravidade de doenças infecciosas pregressas. A história familiar deve constar dados a respeito de consangüinidade dos pais, morte precoce, infecções graves ou repetidas e ocorrência de doenças autoimunes ou neoplasias em parentes próximos ou distantes.
 A história clínica de uma criança com suspeita de imunodeficiência (ID) deve abranger dados como doença materna, duração da gestação, peso e condições do nascimento, relato da queda do coto umbilical (> 21 dias), a natureza e a gravidade de doenças infecciosas pregressas. A história familiar deve constar dados a respeito de consangüinidade dos pais, morte precoce, infecções graves ou repetidas e ocorrência de doenças autoimunes ou neoplasias em parentes próximos ou distantes.")
67
Síndrome da Imunodeficiência Combinada Grave ligada ao Cromossomo X
Etiologia É a imunodeficiência combinada grave (SCID) mais comum, com incidência maior que 1: nascidos vivos. Trata-se de uma herança ligada ao X com completa ausência dos linfócitos T e células NK, mas com desenvolvimento preservado dos linfócitos B. A doença é causada por mutações no gene que codifica a cadeia gama comum do receptor da Interleucina 2 (IL2 RG) localizado no cromossomo X, porém vários estudos têm demonstrado que a cadeia gama não é somente um membro do IL2 R, mas também de outras Interleucinas como: IL4, IL7, IL9, IL15 e IL21.
 mais comum, com incidência maior que 1: nascidos vivos. Trata-se de uma herança ligada ao X com completa ausência dos linfócitos T e células NK, mas com desenvolvimento preservado dos linfócitos B. A doença é causada por mutações no gene que codifica a cadeia gama comum do receptor da Interleucina 2 (IL2 RG) localizado no cromossomo X, porém vários estudos têm demonstrado que a cadeia gama não é somente um membro do IL2 R, mas também de outras Interleucinas como: IL4, IL7, IL9, IL15 e IL21.")
68
Diagnóstico A SCID é assintomática ao nascimento e não pode ser identificada através do exame clínico do RN, porém os sintomas podem surgir precocemente em torno dos três meses de vida, com diarréia crônica, candidíase oral persistente, otite média recorrente, infecção respiratória severa (pneumonia intersticial), eritrodermia, atraso no crescimento e desenvolvimento e septicemia. Essas crianças são particularmente susceptíveis a infecções por patógenos oportunistas, como: Candida albicans, citomegalovírus, pneumocystis carinii e Epstein-Barr, dentre outros. Em crianças normais a contagem de linfócitos pode variar de 2000 a células/mcL, porém, valores abaixo de 4000 células/mcL são consideradas linfopênicos e podem identificar, ao nascimento, as crianças com risco para SCID. Através da citometria de fluxo poderão ser avaliados os subtipos de linfócitos, onde se encontra a ausência de células T e Natural Kiler, mas valores normais ou elevados de células B. Apesar do número de células B estarem normais, os níveis séricos de imunoglobulinas como IgG e IgA encontram-se baixos ou indetectáveis e a resposta anticórpica específica ausente. Observa-se ausência de resposta proliferativa a mitógenos, como a fito-hemglutinina, e a antígenos como o toxóide tetânico. Durante os primeiros 5 a 6 meses de vida, o diagnóstico de SCID pode ser difícil de ser estabelecido devido à presença da IgG materna. Há atrofia do tecido linfóide e hipoplasia tímica (ausência da sombra tímica ao RX de tórax), sendo a biopsia do tecido linfóide raramente necessária para se estabelecer o diagnóstico.
, eritrodermia, atraso no crescimento e desenvolvimento e septicemia. Essas crianças são particularmente susceptíveis a infecções por patógenos oportunistas, como: Candida albicans, citomegalovírus, pneumocystis carinii e Epstein-Barr, dentre outros. Em crianças normais a contagem de linfócitos pode variar de 2000 a células/mcL, porém, valores abaixo de 4000 células/mcL são consideradas linfopênicos e podem identificar, ao nascimento, as crianças com risco para SCID. Através da citometria de fluxo poderão ser avaliados os subtipos de linfócitos, onde se encontra a ausência de células T e Natural Kiler, mas valores normais ou elevados de células B. Apesar do número de células B estarem normais, os níveis séricos de imunoglobulinas como IgG e IgA encontram-se baixos ou indetectáveis e a resposta anticórpica específica ausente. Observa-se ausência de resposta proliferativa a mitógenos, como a fito-hemglutinina, e a antígenos como o toxóide tetânico. Durante os primeiros 5 a 6 meses de vida, o diagnóstico de SCID pode ser difícil de ser estabelecido devido à presença da IgG materna. Há atrofia do tecido linfóide e hipoplasia tímica (ausência da sombra tímica ao RX de tórax), sendo a biopsia do tecido linfóide raramente necessária para se estabelecer o diagnóstico.")
69
Manejo e Tratamento A imunização com vírus vivo atenuado deve ser evitada, assim como todos os produtos sanguíneos devem ser irradiados antes que a transfusão seja realizada nestes pacientes. Utilizar antibióticos e antifúngicos profilaticamente para evitar infecção e repor a gamaglobulina intravenosa regularmente. Tratar qualquer infecção de forma rápida e agressiva e utilizar nutrição parenteral nos casos de atraso do crescimento e desenvolvimento. O tratamento definitivo consiste no transplante de medula óssea histocompatível e quando realizado nos primeiros três meses de vida apresenta uma expectativa de sobrevida superior a 96%. Por isso, é importantíssimo que o diagnóstico seja realizado precocemente, pois a SCID é considerada uma emergência pediátrica cuja morte freqüentemente ocorre antes do primeiro aniversário. Atualmente a terapia gênica apresenta-se como uma esperança para os pacientes portadores de SCID ou de outras imunodeficiências cuja base molecular seja conhecida.

70
OBRIGADO! OBRIGADO!

Apresentações semelhantes

>")


 Reação mais comum durante a transfusão Motivo:>")














