Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Sessão anatomo-clínica Síndrome de Marfan
Escola Superior de Ciências da Saúde (ESCS/SES/DF Dr. Marcos E.A. Segura(Patologista) Dra. Sueli R. Falcão (Cardiologista) Dr. Paulo R. Margotto (Neonatologista) Dra.Maria Tereza Alves S. Rosa (R3 Genética) Dda.Camila Falcão Dda.André Ribeiro 26/4/2010
 Dra. Sueli R. Falcão (Cardiologista) Dr. Paulo R. Margotto (Neonatologista) Dra.Maria Tereza Alves S. Rosa (R3 Genética) Dda.Camila Falcão. Dda.André Ribeiro. 26/4/2010.")
2
Internação materna CESS, 33 anos, com 31semanas e 4 dias de gestação , G2 C1 A0, último parto há 3 anos, deu entrada no PS de GO no dia 28/06/09 devido à ecocardiograma fetal mostrar insuficiência mitral e tricúspide acentuada. Já havia feito esquema completo de corticóide para amadurecimento pulmonar fetal.

3
Ecografia gestacional
22/06/09: feto cefálico com movimentação ativa BCF:158bpm. Presença de cardiomegalia, imagem sugestiva de diminuição do calibre da artéria pulmonar(alteração das vias de saída). Líquido amniótico com diminuição leve ILA=7. Circunferência abdominal fetal em torno do p10 para IG de 32s. Sugerimos investigar intolerância à carboidratos.
. Líquido amniótico com diminuição leve ILA=7. Circunferência abdominal fetal em torno do p10 para IG de 32s. Sugerimos investigar intolerância à carboidratos.")
4
Ecocardiograma fetal 25/06/09: Feto único, IG 32semanas. Conexões venoatriais, atrioventriculares e ventrículo-arteriais concordantes. Presença de cardiomegalia global. Valva tricúspide espessada com regurgitaçãode grau acentuado. Valva mitral também espessada com regurgitação de grau acentuado. Valvas aórtica e pulmonar trivalvulares com regurgitação discreta. Forame oval patente com fluxo da direita para esquerda. Septo ventricular íntegro. Canal arterial pérvio com fluxo pulmonar-aorta não restritivo. Conclusão: Dados sugestivos de IC fetal. Insuficiência mitral e tricúspide de grau acentuado.

5
Internação materna Cardiologista sugeriu investigar infecção transplacentária e iniciar digoxina. Foi feito digoxina por 12 dias. Devido à piora do quadro cardíaco fetal foi realizada cesareana no dia 10/06/09, sem intercorrências. Sorologias: HIV NR, VDRL NR, hepatite B NR, rubéola NR, toxoplasmose IgG e IgM negativos. Tipo sanguíneo mãe: A+

6
Internação do RN Dados do nascimento: Criança nascida de parto cirúrgico às 10:53 hs do dia 10/07/09 (IG: 31s 4 dias)em apresentação cefálica, vivo, único, sexo feminino,chorou ao nascer porem com choro fraco, cianose central, líquido amniótico claro e cordão com duas artérias e uma veia. Apgar de 7/8, com placenta pesando 525 g.Foi diagnosticado possível rotura alta das membranas 21 dias antes do parto. Reanimado ao nascer e persistiu com desconforto respiratório sendo então entubado.
em apresentação cefálica, vivo, único, sexo feminino,chorou ao nascer porem com choro fraco, cianose central, líquido amniótico claro e cordão com duas artérias e uma veia. Apgar de 7/8, com placenta pesando 525 g.Foi diagnosticado possível rotura alta das membranas 21 dias antes do parto. Reanimado ao nascer e persistiu com desconforto respiratório sendo então entubado.")
7
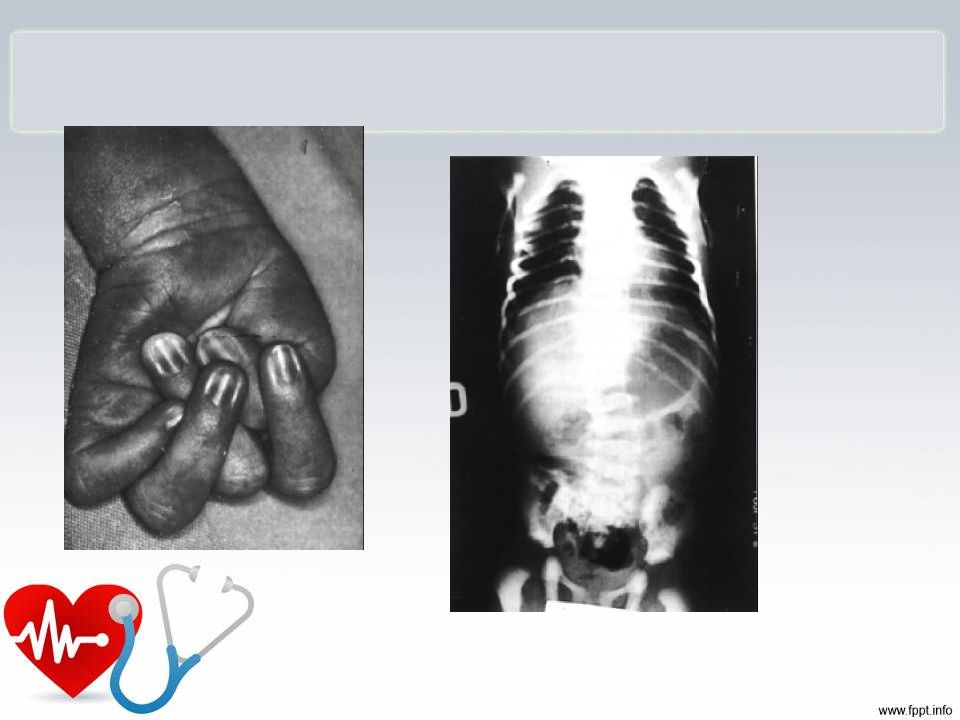
Internação do RN Exame físico ao nascer: Peso:1795g, FC: 160bpm, PC:32 g, estatura de 45cm Paciente em mau estado geral, cianótico, ativo e reativo, pé torto congênito, microftalmia bilateral, micrognatia, baixa implantação de orelhas, dedos dos membros inferiores e superiores alongados e mal posicionados, petéquias em região cefálica. Genitália feminina , anus pérvio.Bulhas hiperfonéticas. Abdome normal, sem visceromegalias. Da sala de parto, após socorros imediatos, o paciente foi para a UTIN.

8
Internação do RN na UTIN
Paciente seguiu grave, hemodinamicamente instável, com taquicardia, baixa perfusão periférica,pulsos centrais regulares e em anuria. Foi feito: Dobutamina 10 Dopamina 5 Fentanil ,Midazolam Furosemida 3 mg/kg/dia VM FiO FR de 50. PEEP 5 Ampicilina + Gentamicina Adrenalina Prostin de 0,02

9
Internação do RN na UTIN
O paciente seguiu grave, apresentando piora hemodinâmica progressiva, mesmo em uso de drogas vasoativas em crescendo, sedado.O esquema antibiótico foi trocado por cefepime e amicacina, com correção da dose pela ira. Foi a óbito às 16 horas do dia 12/07/09 por provável choque cardiogênico agravado com choque séptico.

10
Exames complementares RN
Raio X de tórax: Aumento da área cardíaca, hipofluxo pulmonar mostranto TOT bem posicionado e cateter umbilical em fígado. Gasometria arterial: (VM c/ FiO2 100% PIP de 20 PEEP de 4) pH= pCO2=35.1,pO2=22, HCO3=15.5, BE= -12,6 SatO2= 47% 11/07/09:Hb 12,1 Htc 33% leucócitos (N62B01L32M05E00B00) Na, K, Cl: faltou reagente Uréia 38 creatinina 1.2 calcio 6,3
 pH= pCO2=35.1,pO2=22, HCO3=15.5, BE= -12,6 SatO2= 47% 11/07/09:Hb 12,1 Htc 33% leucócitos (N62B01L32M05E00B00) Na, K, Cl: faltou reagente. Uréia 38 creatinina 1.2 calcio 6,3")
11
Exames complementares RN
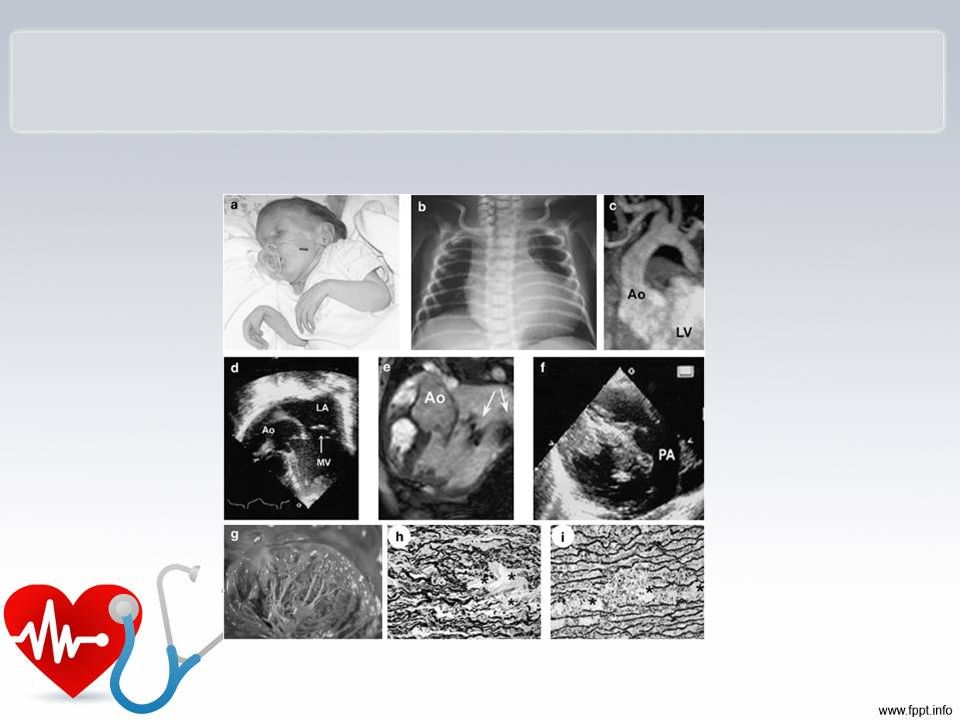
Ecocardiografia : Situs sólitus em bradicardia .Conexões AV e VA concordantes. Dilatação aneurismática do átrio direito e importante em demais câmaraS. Hipocontratilidade difusa de paredes de ambos os ventriculos. CIA em região de fossa oval medindo cerca de 7 mm com shunt bidirecional. Displasia de váculas tricúspide e mitral com abertura normal e má coaptação, apresentando importante refluxo de ambas as válvulas. Válvula aórtica normaL. Válvula pulmonar com abertura diminuída por baixo débito cardíaco e morfologia normal. Refluxo mitral e tricúspide importantes. Sem coarctação aórtica. Canal arterial com 3 mm co fluxo e/d Conclusão: Cardiomiopatia dilatada de ambos os venrtriculos com disfunção sistólica importante. Displasia das válvulas mitral e tricúspide. Com insuficiência importante.Cia tipo fossa oval .Persistência do canal arterial.Fluxo pulmonar dependente do canal arterial devido a disfunção do ventrículo direito.

12
Comentários da Genética
Maria Teresa Alves da Silva Rosa R3 Genética Médica

13
Consulente encaminhada por obstetra
34 anos, tem sopro cardíaco . Marido 42 anos hígido. Não consanguíneos. G2P2A0 G1 há 3 anos, criança faleceu com 1ano e 3 meses por broncopneumonia, edema , hemorragia pulmonares, miocardite e esteatose hepática achados sugestivos de infecção viral G2 há 6 meses, sexo feminino, diagnóstico intra-uterino de cardiomegalia ( feto 31 s + 3d). Criança nasceu com 31 s,peso 1750g. Apresentava algumas dismorfias : micrognatia, microftalmia, aracnodactilia Houve achados de degeneração mixedematosa de válvulas cardíacas, degeneração de fibras elásticas das válvulas cardíacas Em foto foi identificada contratura dos punhos Na história familiar prima da mãe tem cardiopatia congênita Cariótipo do casal normal Ecocardiograma da mãe evidencia discreta insuficiênica mitral
. Criança nasceu com 31 s,peso 1750g. Apresentava algumas dismorfias : micrognatia, microftalmia, aracnodactilia. Houve achados de degeneração mixedematosa de válvulas cardíacas, degeneração de fibras elásticas das válvulas cardíacas. Em foto foi identificada contratura dos punhos. Na história familiar prima da mãe tem cardiopatia congênita. Cariótipo do casal normal. Ecocardiograma da mãe evidencia discreta insuficiênica mitral.")
14
Síndrome da Aracnodactilia Contratual de Beals

15
Características Membros longos e delgados, com aracnodactilia, campodactilia, desvio ulnar dos quirodáctilos, contraturas articulares, principalmente dos joelhos e quadris Cifoescoliose, pescoço relativamente curto, metatarso varo, pé equinovaro, hipoplasia dos músculos da panturrilha Orelhas com aparência amarrotada com as conchas maldefinidas e ramo proeminente da raiz da hélice

16
Achados eventuais Micrognatia Anomalias cranianas
Coloboma de íris ceratocone, miopia Pectus escavatum, carinatum Subluxação da patela Defeito septal atrial, defeito septal ventricular, dilatação da raiz da aorta, prolapso da valva mitral

17
Etiologia Autossômica dominante com penetrância incompleta, expressividade variável Lócus da fibrilina:cromossomo 5q23-31(FBN2)
 .")
18
Forma Severa Forma grave, letal no período neonatal
Graves defeitos cardíacos incluindo arco aórtico interrompido, defeito septal atrial e dilatação da raiz da aorta Anomalias gastrintestinais: atresia esofágica e duodenal, má rotação intestinal

21
Síndrome de Marfan

22
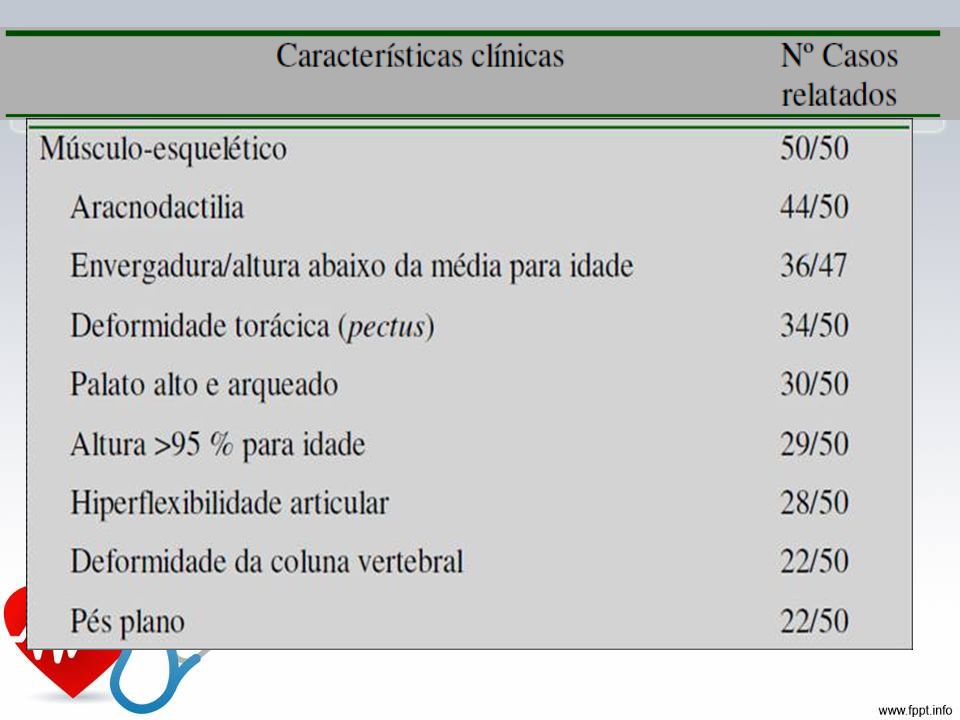
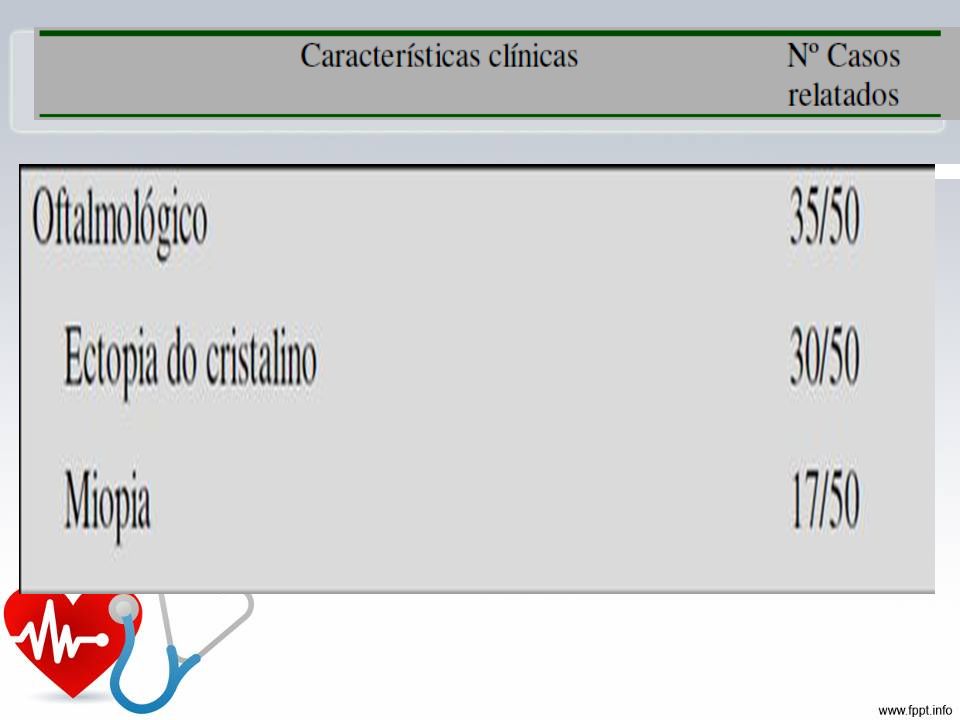
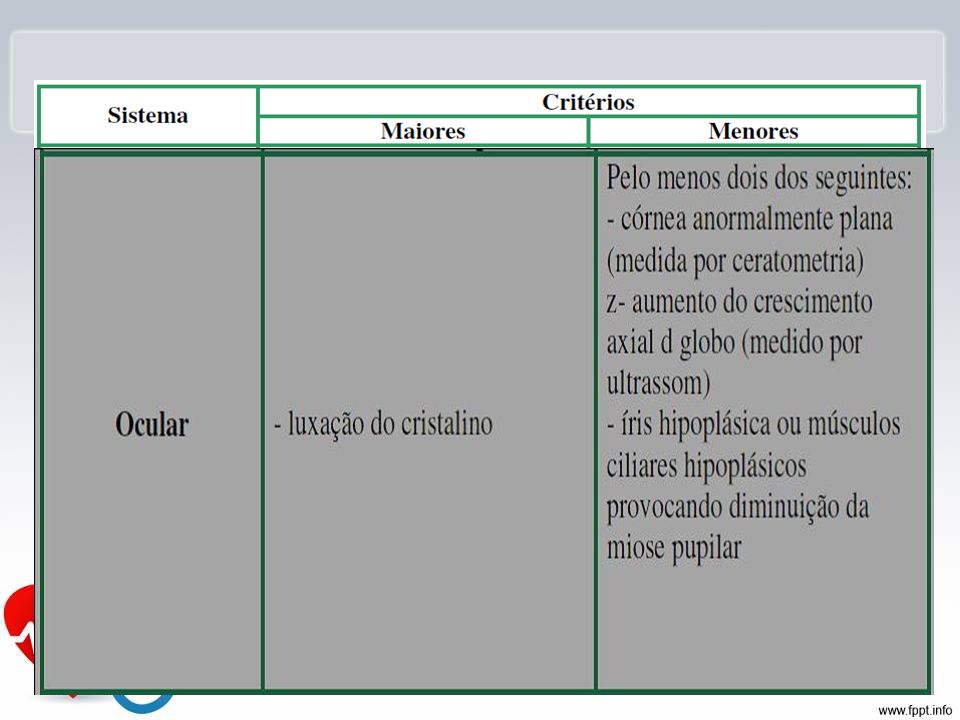
Características Crescimento: tendência em desenvolver alta estatura, com longos e alongados, pouca gordura subcutânea, hipotonia muscular Esqueléticas: pectus carinatum, pectus escavatum, proporção diminuída entre o segmento superior e inferior, envergadura maior que estatura, sinal do polegar e do punho, pés planos, acetábulo protuso. Oculares: subluxação do cristalino, normalmente superior, com defeito de ligamento suspensor, córnea plana, comprimento axial do globo ocular aumentado, íris .

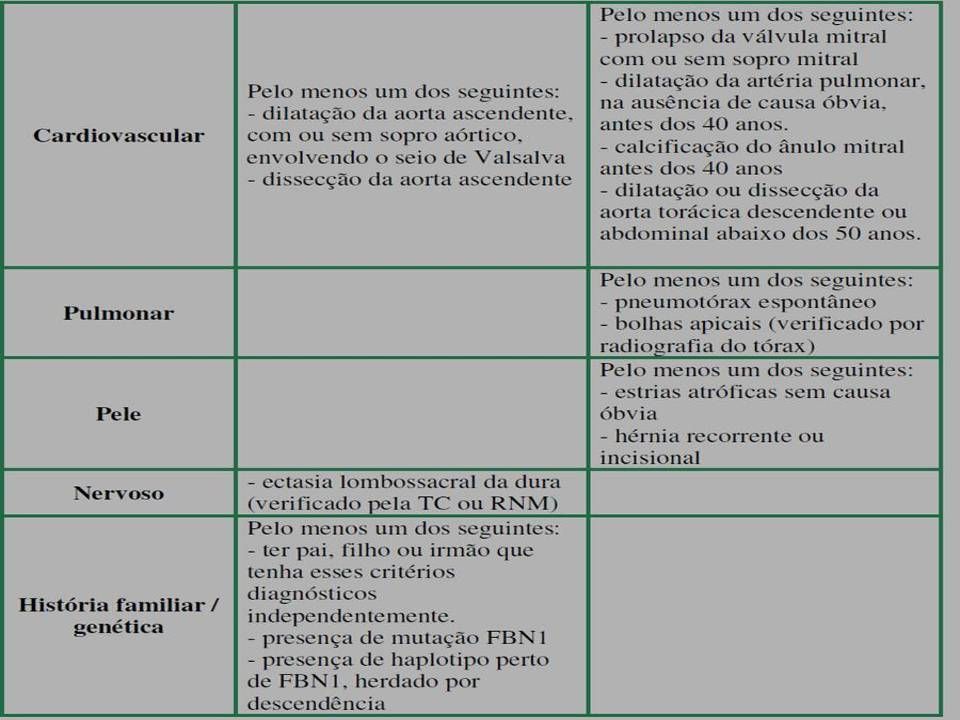
23
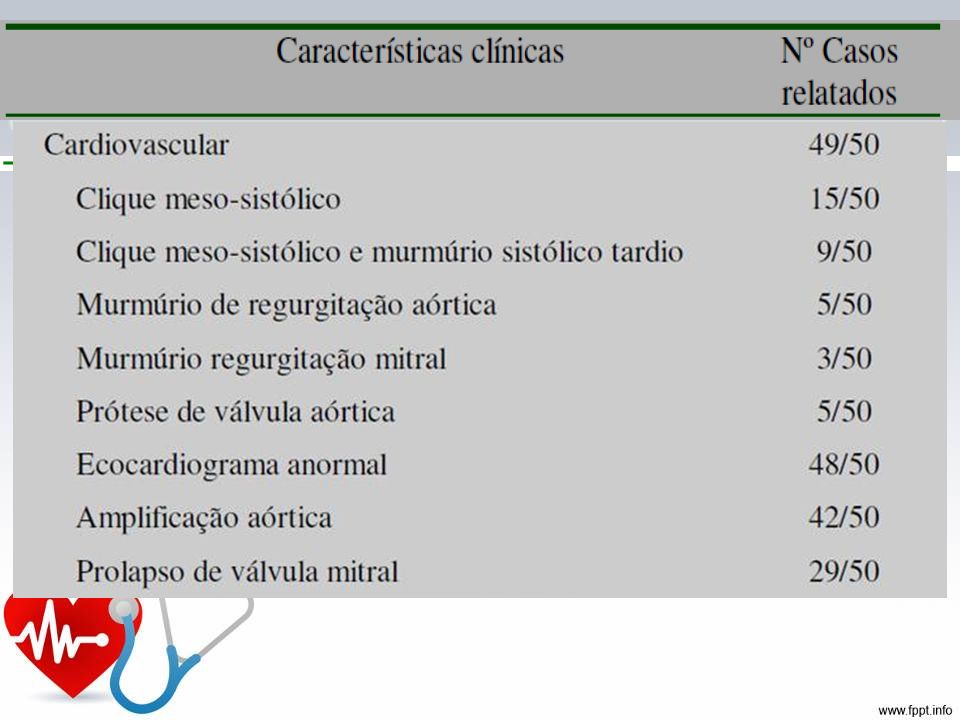
Características Cardiovasculares: dilatação da aorta ascendente, com ou sem regurgitação aórtica, dissecção da aorta ascendente, prolapso de válvula mitral, dilatação da artéria pulmonar, dilatação ou dissecção da aorta abdominal ou torácica descendente Pulmonares: pneumotórax espontâneo, bollhas apicais Pele e revestimento: ectasia dural, lombosacra

24
Etiologia Autossômica dominante, com expressividade variável
Mutação do gene da fibrilina localizado no cromossomo 15q Fibrilina é uma glicoproteína que é componente das microfibrilas extracelulares que são os principais componentes das fibras elásticas

25
Forma Severa Síndrome de Marfan Grave nos primeiros 3 meses de vida
Graves defeitos cardíacos, incluindo prolapso da válvula mitral, regurgitação valvular e dilataçào da raiz da aorta, contraturas congênitas podem estar presentes Dolicocefalia, palato arqueado, micrognatia, articulações hiperextensíveis, aracnodactilia, pés planos, deformidade torácica, iridonese, megalocórnea, luxação do cristalino 14% das crianças falecem antes do 1o ano de vida

27
Conclusão Criança apresentava algumas dismorfias: retrognatia, aracnodactilia, contraturas articulares Achados compatíveis com síndrome Marfan ou Beals Síndromes semelhantes, podem fazer parte de um espectro de variações fenotípicas com o mesmo defeito genético fundamental Como criança não foi avaliada pela genética, não é possível afirmar com certeza tratar-se de Sd. Marfan ou outra síndrome Nesse caso pode tratar-se de um acontecimento esporádico ou uma doença herdada Se esporádico risco de recorrência baixo, se herdado risco de recorrência de 50% Casal deverá retornar à genética em caso de outra gestação

28
Considerações Importância da necropsia para diagnóstico
Importância de trabalho multidisciplinar Valorizar o diagnóstico

29
Discussão dos Ddos André Ribeiro e Camila Falcão
SÍNDROME DE MARFAN

30
INTRODUÇÃO Ocorre devido a mutações do cromossomo 15.
Variados graus de mutação do gene FBN-1, acarretando variedades na apresentação. Hereditariedade marcante. É uma doença do tecido conjuntivo, que afeta principalmente: Músculo-esquelético Ocular Cardiovascular (boa parte dos óbitos)
")
31
Histórico 1896: Bernard Marfan descreveu o protótipo da síndrome.
1931: Weve-Síndrome da distrofia mesodermalis congenita Vários personagens da história e do esporte possuiam a síndrome.

36
Epidemiologia Prevalência recente: 10 : 100.000
Aumento progressivo na expectativa de vida do paciente.

37
Fibrilina Componete fundamental da MEC
Sintetizados por várias células da MEC, principalmente os fibroblastos. Localiza-se em todas as interfaces epitélio-mesenquimais do corpo: Artérias elásticas Oculares Tecido ósseo e articular

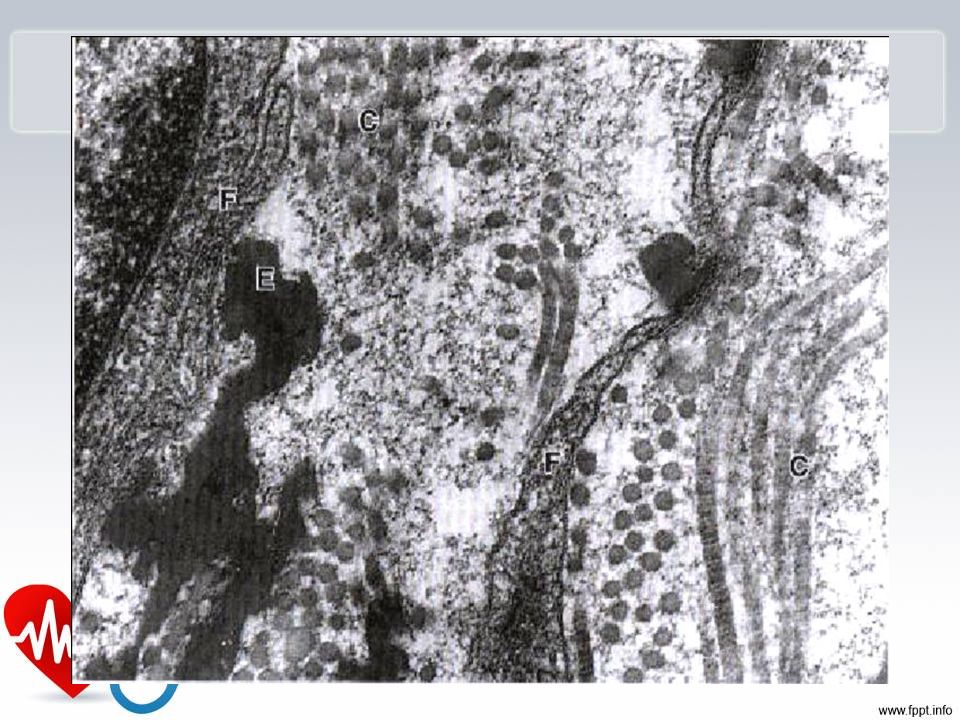
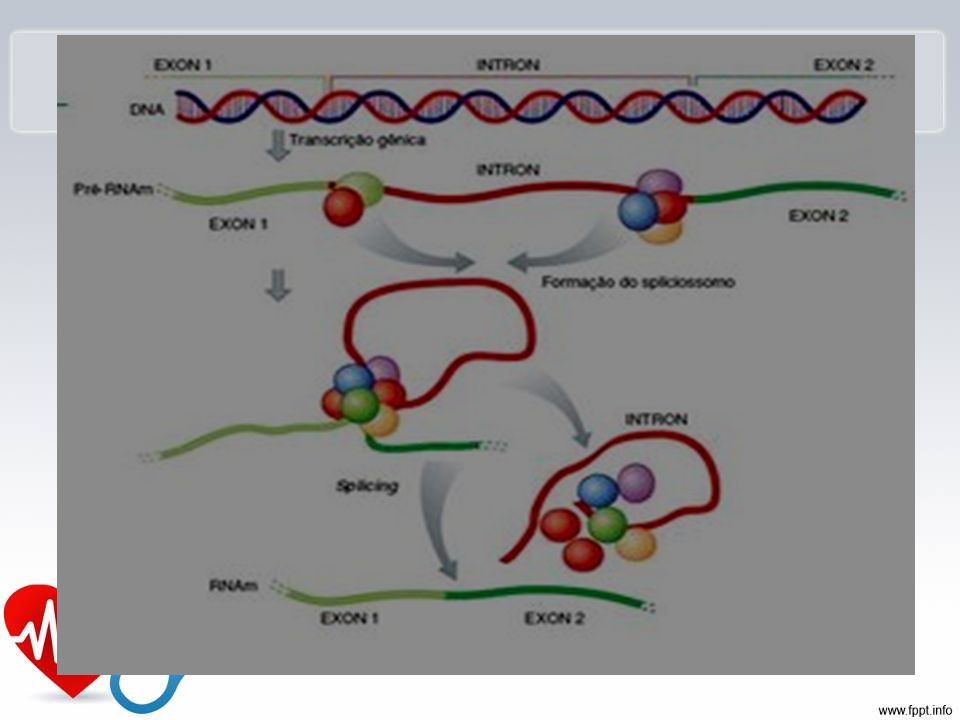
40
Etapas de formação

41
Fibrilina

47
Sinal de Steinberg

48
Sinal de Murdoch

49
Pectus

50
Pes planus

51
Escoliose

52
Ectopia do cristalino

53
Outros sistemas afetados
Pele Sistema nervoso e dura máter Ossos Pulmões

54
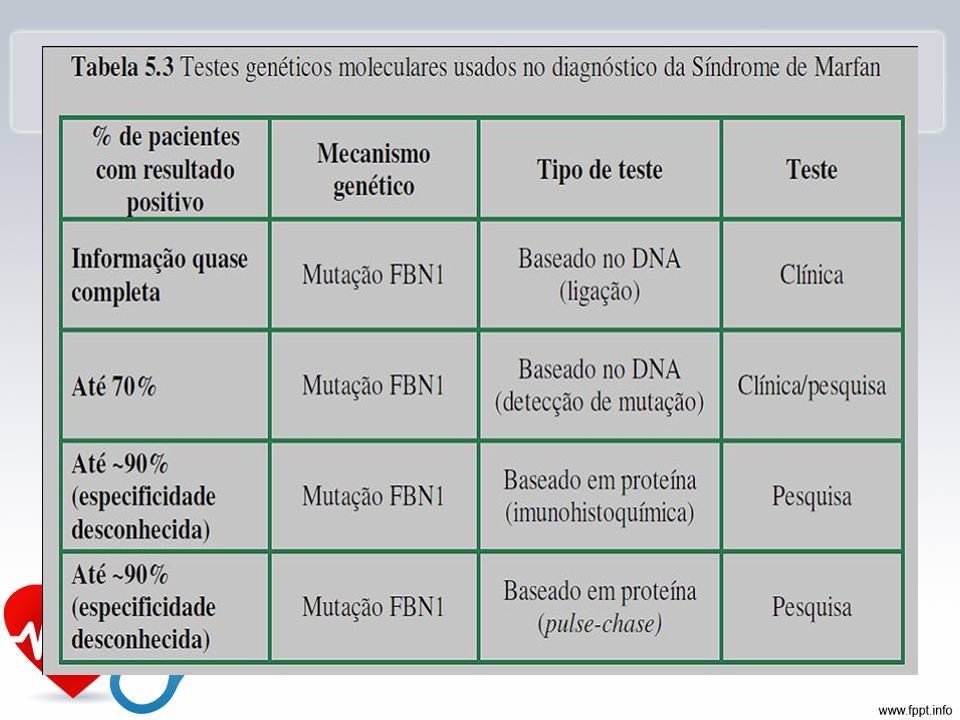
Diagnóstico: Critérios maiores em dois sistemas e em um terceiro
Clínico:

58
Diagnóstico diferencial
Homocistinúria??? Biotipo marfanóide + Ateromatose+ Luxação do cristalino ACC??? Evolução benigna Weil-marchesani??? ectopia do cristalino + dedos roliços e curtos Sindrome de Stikler??? Aracnodactilia + Microftalmia/micognatia + Membros longos + oculares Sindrome de ehler-danlos: Acometimento vascular + anormalidades articulares + pele laxa

59
Insuficiência cardíaca dilatada
São doenças miocárdicas atribuídas a causas além das valvulares, coronarianas, pericárdicas ou por cardiopatias congênitas Alterações nos miócitos, que em certo momento apresentará erros funcionais Podem ser decorrentes de doenças herdadas ou por infeccções ou arritmias.

60
Predisposição para ICC
Anormalidades mitocondriais Metabolismo de ácidos graxos Anormalidades protéicas nos cardiomiócitos Toxinas e infecções sépticas

61
Manejo clínico Descoberta precoce: Aspecto fundamental
Aconselhamento genético Requer a participação multidisciplinar: Geneticista Oftalmologista Ortopedista Cirurgião cardiovascular

62
Bibliografia EMMANOUILIDES, G. C.; RIEMENSCHNEIDER, T. A.; ALLEN, H. D.; GUTGESELL, H. P. Heart disease in infants, children, and adolescents, Vol II. 15th ed. Willians & Wilkins Editors: Baltimore KUMAR, V. K.; ABBAS, A. K.; FAUSTO, N. Patologia – Bases Patológicas das doenças. 7ª ed. Elsevier Editora: Rio de Janeiro JUNQUEIRA, L. C.; CARNEIRO, J. Histologia Básica. 10ª ed. Guanabara Koogan Editora: Rio de Janeiro BARRETO, M. M.; BRESSANE, R. C.; MENGUER, R. K.; SILVEIRA, S. M.; ALBERTI, T. Z.; MALDOTTI, V. DUPRAT, A. C.; PEREIRA, C. Síndrome de Marfan. (?) ed. Publicação do Departamento de Ciências Morfológicas. Porto Alegre
 ed. Publicação do Departamento de Ciências Morfológicas. Porto Alegre")
63
Comentários da PatologiaA 022 / 10
Dr. Marcos E.A. Segura médico patologista

64
Ectoscopia RN, sexo feminino Cianótico Micrognatia, microftalmia
Quirodáctilos e pododáctilos alongados Cateter em jugular direita e umbilical

65
Ectoscopia RN, sexo feminino Cianótico Micrognatia, microftalmia
Quirodáctilos e pododáctilos alongados Cateter em jugular direita e umbilical

66
Ectoscopia Referência Peso 1750,0g C.Total 46,0cm C.Cr-Cau 32,0cm
C.Cef 30,0cm C.Tor 26,0cm C.Abd Cérebro 270,0g Coração 27,0g Pulmão 40,0g Figado 83,0g Baço - Adrenal Rim 14,0g

67
Sistema Cardiovascular
Cardiomegalia Cateter localizado em AD

68
Sistema Cardiovascular

69
Sistema Cardiovascular

70
Sistema Cardiovascular

71
Sistema Cardiovascular

72
Sistema Cardiovascular
Miocárdio com alterações discretas relacionadas a hipertrofia Histologia do miocárdio

73
Sistema Cardiovascular – Válvulas AV
Válvulas Cardíacas Espessadas Histologia das Válvulas

74
Depósito de material intersticial mixomatoso
Sistema Cardiovascular– Válvulas AV Depósito de material intersticial mixomatoso

75
Depósito de material intersticial mixomatoso
Sistema Cardiovascular – Válvulas AV Depósito de material intersticial mixomatoso

76
Área de degeneração cística da camada média de artéria pulmonar
Sistema Cardiovascular – Artéria Pulmonar Área de degeneração cística da camada média de artéria pulmonar

77
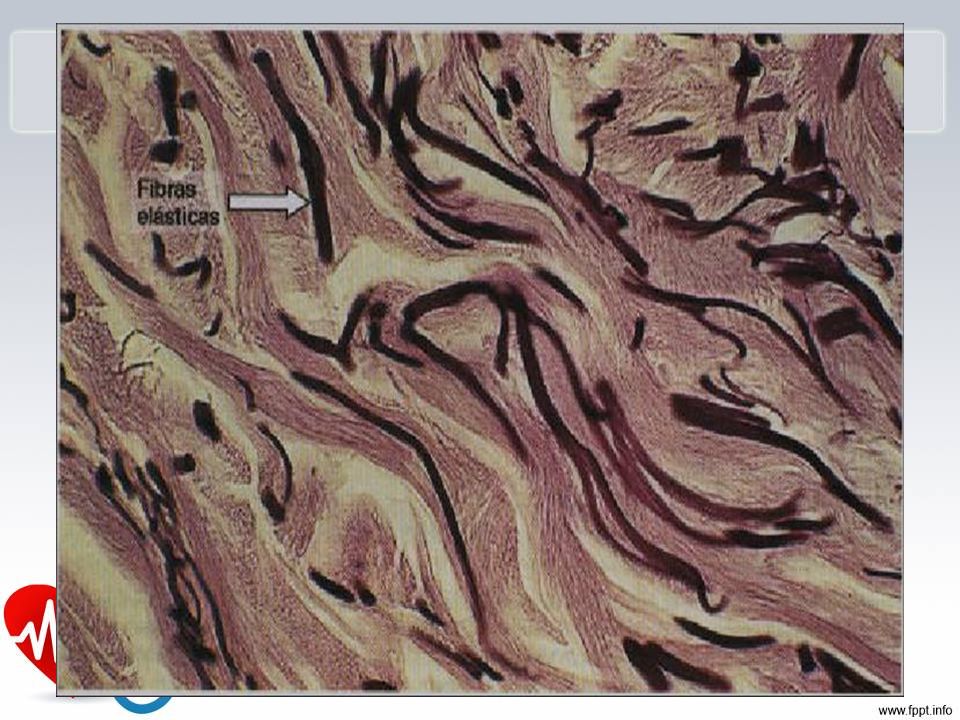
Sistema Cardiovascular – Artéria Pulmonar
COLORAÇÃO PARA FIBRAS ELÁSTICAS: Camada média de artéria pulmonar demonstra interrupção das fibras elásticas formando área pseudo-cística com depósito de substância intersticial (Necrose Cística da Camada Média)
")
78
Sistema Cardiovascular – Aorta

79
SÍNDROME DE MARFAN Sistema Cardiovascular Conclusões Cardiomegalia
Degeneração mixomatosa de válvulas cardíacas Necrose cística da média de artéria pulmonar SÍNDROME DE MARFAN

80
Sistema Respiratório Traquéia e brônquios com pus na luz
Pulmões com áreas de consolidação (hepatização)
")
81
Sistema Respiratório

82
Sistema Respiratório

83
Sistema Respiratório Conclusões Pneumonia Doença da membrana hialina

84
Sistema Digestivo Trato Gastrointestinal Esôfago pérvio
Rotação intestinal habitual

85
Fígado

86
Baço

87
Conclusões Fígado Baço Congestão passiva (ICC) Colestase
Hiperplasia da polpa branca (infecção/sepse)
")
88
Sistema Urinário

89
Sistema Urinário Conclusões Imaturidade do parênquima renal

90
Encéfalo

91
Encéfalo

92
Conclusões – Causa mortis e Doença Principal
Síndrome de Marfan Prematuridade Degeneração Mixomatosa de válvulas AV Pneumonia bilateral Doença da Membrana Hialina Cardiomegalia Septicemia Insuficiência Respiratória Choque Cardiogênico Congestão Passiva Crônica em Fígado e baço

93
Preenchimento da Declaração de Óbito
Estado / Família Fim da vida civil de um cidadão Secretaria de Saúde Informa as circunstâncias da morte Estatística Tomada de decisões

94
Preenchimento da Declaração de Óbito
Preenchimento obrigatório pelo médico assistente Não pode se recusar Não pode cobrar NÃO PREENCHER A DECLARAÇÃO EM CASO DE MORTE EXTERNA

95
Preenchimento da Declaração de Óbito
Parte I : Cartório Parte II : Identificação Parte III : Residência Parte IV : Ocorrência Parte V : Óbito fetal ou menor que 1 ano Parte VI : Condições e causas do óbito Parte VII : Identificação do Médico Parte VIII : Causas Externas Parte IX : Localidades sem Médicos

96
Preenchimento da Declaração de Óbito
Choque Cardiogênico Valvulopatia Cardíaca Síndrome de Marfan X Prematuridade Pneumonia

97
Síndrome de Marfan Doença do tecido conjuntivo Prevalência 1:5000
70-85% familiar (autossômica dominante) Aconselhamento genético Patogênese Defeito em um glicoproteína (fibrilina-1) Fibras elásticas
 Aconselhamento genético. Patogênese. Defeito em um glicoproteína (fibrilina-1) Fibras elásticas.")
98
Síndrome de Marfan Morfologia Esqueleto Altos Extremidades longas

99
Síndrome de Marfan Morfologia Cardiovascular Ocular
Prolapso da Válvula Mitral Dilatação da aorta (necrose cística da média) Ocular Ectopia de cristalino
 Ocular. Ectopia de cristalino.")
100
HOSPITAL REGIONAL DA ASA SUL/SES/DF

101
INTERNATO NA ESCS: VOCÊ FAZ A DIFERENÇA!
TURMA-2005-ESCS-GRUPO E INTERNATO NA ESCS: VOCÊ FAZ A DIFERENÇA!

Apresentações semelhantes




















 MSS 22 anos, procura o serviço para fazer um cariótipo e receber aconselhamento genético. È G1, P1, A0, teve um filho pré termo.>")



