Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Síndromes Mieloproliferativas Crônicas
Diagnóstico, prognóstico e tratamento Camila Linardi 2008

2
Síndromes Mieloproliferativas Crônicas
Doenças clonais da célula tronco hematopoética Trombocitemia Essencial Policitemia Rubra Vera Mielofibrose Primária LMC

3
Policitemia Rubra Vera
Incidência 1-2.3/ ; Idade mediana: 60 anos; ♂ > ♀; 80% assintomáticos, 20% com trombose; Sobrevida global: 18 meses se não tratado; Sobrevida global com tratamento: > 15 anos; MF: 10-30% em anos; SMD/LMA: 1,5-15,0%

4
Diagnóstico: 2 primeiros maiores e mais 1 maior ou 2 menores
Policitemia Vera - Critérios Diagnósticos OMS Maiores Massa eritrocitária > 25% do normal ou Hb > 18,5 (h) e 16,5 (m) Ausência de causas de poliglobulia secundária: ausência de eritrocitose familiar ausência de aumento de EPO (hipóxia, EPO exógena, receptor de EPO truncado, Hb alta afinidade, tumor produtor de EPO) Esplenomegalia palpável Cariótipo Anormal (excluindo Ph) (~20% dos casos→+8,+9, del20q, del13q, del1p) Formação de colônia eritroide endógena in vitro Menores Plaquetas > /mm3 Leucócitos > /mm3 (fumante > /mm3) Bx MO com panmielose EPO sérica diminuída Diagnóstico: 2 primeiros maiores e mais 1 maior ou 2 menores
 e 16,5 (m) Ausência de causas de poliglobulia secundária: ausência de eritrocitose familiar. ausência de aumento de EPO (hipóxia, EPO exógena, receptor de EPO truncado, Hb alta afinidade, tumor produtor de EPO) Esplenomegalia palpável. Cariótipo Anormal (excluindo Ph) (~20% dos casos→+8,+9, del20q, del13q, del1p) Formação de colônia eritroide endógena in vitro. Menores. Plaquetas > /mm3. Leucócitos > /mm3 (fumante > /mm3) Bx MO com panmielose. EPO sérica diminuída. Diagnóstico: 2 primeiros maiores e mais 1 maior ou 2 menores.")
5
Policitemia Vera- Novos Critérios Diagnósticos OMS-2008
Critérios Maiores: Hb>18,5mg/dl em ♂ ou Hb>16,5mg/dl em ♀ ou ↑volume eritrocitário; Mutação JAK 2 Critérios Menores: Biopsia MO característica; EPO↓; Formação de colônia eritroide endógena in vitro. Diagnóstico: 1o maior e 2 menores ou 2 maiores e 1 menor

6
Policitemia Rubra Vera
Tratamento visa diminuir risco de trombose com menor risco possível de LMA; Modalidades mais usadas: flebotomia, quimioterapia, IFN; Poucos trabalhos randomizados.

7
Policitemia Rubra Vera
AAS 100mg: estudo randomizado: ↓ morte por causas cardiovasculares e ↓ eventos trombóticos não fatais; Alopurinol: Ácido úrico↑ → alto turnover celular ; Prurido: tratar com antihistamínicos; Sangria: Ht↓ 45% (♂)/ Ht↓ ≤42% (♀); Controle da plaquetose: ↓trombose? Tratar HAS, tabagismo, DM e DLP.
/ Ht↓ ≤42% (♀); Controle da plaquetose: ↓trombose Tratar HAS, tabagismo, DM e DLP.")
8
Policitemia Rubra Vera
Fatores de risco para trombose Idade > 60 anos (incidência trombose 4/100 pacientes-ano) Idade > 70 anos (5.1/100 pacientes-ano) História de trombose prévia Fatores de risco para LMA Quimioterapia
 Idade > 70 anos (5.1/100 pacientes-ano) História de trombose prévia. Fatores de risco para LMA. Quimioterapia.")
9
Policitemia Rubra Vera
Grupos de Risco para Tratamento Baixo risco Idade < 60 anos E Sem história de trombose E Sem fator de risco cardiovascular Risco intermediário Não baixo nem alto Alto Risco Idade >/= 60 anos OU História de trombose prévia

10
Policitemia Rubra Vera
Tratamento baseado em grupos de risco Idade < 60 anos Baixo risco: Flebotomia para Htc < 45% (h) e 43% (m) Risco intermediário: Flebotomia - considerar citorredução se alta necessidade de flebotomia, esplenomegalia progressiva, plaquetas > /mm3 Alto risco: citorredução ( Hidroxiuréia ou IFN )
 e 43% (m) Risco intermediário: Flebotomia - considerar citorredução se alta necessidade de flebotomia, esplenomegalia progressiva, plaquetas > /mm3. Alto risco: citorredução ( Hidroxiuréia ou IFN )")
11
Policitemia Rubra Vera
Tratamento baseado em grupos de risco Idade > 60 anos Todos são de alto risco; Citorredução com Hidroxiuréia para manter Htc < 45% e plaquetas < /mm3; Em pacientes com expectativa de vida < 10 anos considerar Bussulfan ou P32.

12
Trombocitemia Essencial
Incidência 0,6 a 2,5/ ; Mediana de idade: 60 anos; 57%: assintomáticos ao diagnóstico; 29%: sintomas vasomotores; 6%: sangramento; 18% de trombose; Sobrevida em 10 anos = população normal; Morbidade por trombose ou sangramento; 5%: risco de LMA ou MF.

13
Trombocitemia essencial - Critérios Diagnósticos OMS
Critérios Positivos Plaquetas ≥ /mm3 Bx MO com proliferação preferencial de megacariócitos maduros Critérios Negativos Sem evidência de Policitemia Vera: Massa eritrocitária normal ou Hb < 18,5 (♂) e 16,5 (♀) Ferro presente na MO, VCM normal ou ferritina sérica normal Se ferro ausente: prova terapêutica com ferro não eleva o Hb em níveis de PV Ausência de cromossomo Ph ou BCR-ABL Ausência de fibrose colágena ou presença mínima de fibrose reticulínica na MO Sem evidência de alterações morfológicas e citogenéticas sugestivas de SMD Sem causas de trombocitose reativa (inflamação, infecção, neoplasia ou esplenectomia)
 e 16,5 (♀) Ferro presente na MO, VCM normal ou ferritina sérica normal. Se ferro ausente: prova terapêutica com ferro não eleva o Hb em níveis de PV. Ausência de cromossomo Ph ou BCR-ABL. Ausência de fibrose colágena ou presença mínima de fibrose reticulínica na MO. Sem evidência de alterações morfológicas e citogenéticas sugestivas de SMD. Sem causas de trombocitose reativa (inflamação, infecção, neoplasia ou esplenectomia)")
14
Trombocitemia essencial – Novos Critérios Diagnósticos OMS-2008
Critérios Positivos Plaquetas ≥ /mm3 Bx MO com proliferação preferencial de megacariócitos maduros Mutação JAK2 ou outra alteração clonal (exceto BCR-ABL ou Ph) Critérios Negativos Sem evidência de Policitemia Vera Sem evidência de Mielofibrose Sem evidência de LMC Sem evidência de SMD JAK2-: Sem causas de trombocitose reativa (inflamação, infecção, neoplasia ou esplenectomia)
 Critérios Negativos. Sem evidência de Policitemia Vera. Sem evidência de Mielofibrose. Sem evidência de LMC. Sem evidência de SMD. JAK2-: Sem causas de trombocitose reativa (inflamação, infecção, neoplasia ou esplenectomia)")
15
Trombocitemia Essencial
Fatores de risco para trombose Idade > 60 anos Trombose prévia Tabagismo Hipercolesterolemia

16
Trombocitemia Essencial
Fatores de risco para sangramento Plaquetas > /mm3 DvW adquirida Fatores de risco para LMA Anormalidades citogenéticas Tratamento quimioterápico

17
Trombocitemia Essencial
Tratamento visa diminuir risco trombótico Agentes mais usados: Hidroxiuréia, Anagrelide, IFN, AAS Apenas um estudo randomizado comparando Hidroxiuréia e observação: Hidroxiureia: eventos trombóticos <4% Observação: eventos trombóticos: 24% Anagrelide : estudo prospectivo randomizado comparou com Hidroxiurea : ↑ trombose arterial, ↓ trombose venosa, ↑Hemorragia e ↑risco de morte decorrente de trombose. Não há trabalhos randomizados em pacientes de baixo risco para trombose

18
Trombocitemia Essencial
Grupos de risco para Tratamento Baixo risco Idade < 60 anos E Sem história de trombose E Plaquetas < /mm3 E Sem fator de risco cardiovascular (tabagismo/colesterol) Risco intermediário Não baixo nem alto Alto Risco Idade >/= 60 anos OU História de trombose prévia
 Risco intermediário. Não baixo nem alto. Alto Risco. Idade >/= 60 anos OU. História de trombose prévia.")
19
Trombocitemia Essencial
Tratamento baseado em grupos de risco Idade < 60 anos Baixo risco: sem tratamento ou AAS; Risco intermediário: sem tratamento ou AAS ou citorredução; Alto risco: citorredução ( Hidroxiuréia ou anagrelide ou INF ); Anagrelide ( versus Hidroxiuréia): ↑trombose arterial; ↑sangramento; ↑fibrose medular; ↓trombose venosa.
; Anagrelide ( versus Hidroxiuréia): ↑trombose arterial; ↑sangramento; ↑fibrose medular; ↓trombose venosa.")
20
Trombocitemia Essencial
Tratamento baseado em grupos de risco Idade > 60 anos Todos são de alto risco Citorredução com Hidroxiuréia para manter plaquetas < /m3 Em pacientes com expectativa de vida < 10 anos considerar uso de P32

21
Principais toxicidades das drogas
PV e TE Principais toxicidades das drogas Hidroxiuréia: citopenias, ulceras orais, diarréia, risco de leucemia aguda? Anagrelide: cefaléia, taquicardia, edema, ICC, anemia, fibrose pulmonar, não relatado risco de leucemia aguda IFN: citopenias, depressão, fadiga, sintomas gripais, disfunção tireoideana, aparentemente sem risco de leucemia aguda P32: citopenias prolongadas, náusea, diarréia, risco de leucemia aguda 2-5% em 7-10 anos

22
Mielofibrose Primária
Incidência / ; Idade mediana anos; 30 % assintomáticos; Sintomas constitucionais: 40 %; Anemia, esplenomegalia e manifestações auto-imunes Mediana de sobrevida: anos; Mortalidade: falência medular, hipertensão portal e LMA ( 5-30%); Mielofibrose secundária: 25 a 50% em PV e 2 a 3% em TE; Citogenética: ~50% dos casos ( del13q, del20q, +1q, +8, anormalidades do 7, 9 e 12, der 6 ).
; Mielofibrose secundária: 25 a 50% em PV e 2 a 3% em TE; Citogenética: ~50% dos casos ( del13q, del20q, +1q, +8, anormalidades do 7, 9 e 12, der 6 ).")
23
Mielofibrose Primária - Critérios Diagnósticos PVSG
Maiores Fibrose medular difusa Ausência de Ph ou BCR-ABL Menores Esplenomegalia Anisopoiquilocitose com hemácias em lágrima Presença de células mielóides imaturas em SP Presença de eritroblastos em SP Dismegacariopoese ou agrupamentos anômalos de megacariócitos na MO Metaplasia mielóide Diagnóstico: 2 maiores + 2 menores se esplenomegalia ou 2 maiores + 4 menores se ausência de esplenomegalia

24
Mielofibrose Primária: Critérios Diagnósticos OMS-2008
Critérios Maiores: Biópsia MO característica; Anormalidade Clonal: JAK2V617F; MPLW515R/K ( 5% dos pacientes com MF); Outra; Sem critérios para PV, LMC ou SMD; Se ausência de anormalidade clonal→ sem evidência de estado reativo; Critérios Menores: Leucoeritroblastose; DHL↑, Anemia, Esplenomegalia. Diagnóstico: todos os critérios maiores e 2 menores.
; Outra; Sem critérios para PV, LMC ou SMD; Se ausência de anormalidade clonal→ sem evidência de estado reativo; Critérios Menores: Leucoeritroblastose; DHL↑, Anemia, Esplenomegalia. Diagnóstico: todos os critérios maiores e 2 menores.")
25
Mielofibrose Primária
Fatores de risco para mortalidade Grau de anemia Leucocitose ou leucopenia Idade > 55 anos Cariótipo anormal Sintomas sistêmicos Monocitose Grau de fibrose medular

26
Mielofibrose Primária
Escore Lille Fatores de risco: Hb < 10 g/dL; Leucócitos < 4000/mm3 ou > /mm3. Grupos de Risco Baixo: 0 fatores (sobrevida 93 meses); Intermediário: 1 fator (26 meses); Alto: 2 fatores (13 meses).
; Intermediário: 1 fator (26 meses); Alto: 2 fatores (13 meses).")
27
Mielofibrose Primária
Escore Cervantes Fatores de risco: Hb < 10 g/dL; presença de ≥ 1% de blastos em SP; sintomas constitucionais. Grupos de Risco Baixo: 0 ou 1 fator (mediana sobrevida = 176 meses); Alto: 2 ou mais fatores (mediana sobrevida = 33 meses).
; Alto: 2 ou mais fatores (mediana sobrevida = 33 meses).")
28
Mielofibrose Primária
Nenhum tratamento convencional mostrou aumento da sobrevida Não há evidência de diminuição do risco de progressão com tratamento convencional Poucos trabalhos com TMO somente com pacientes jovens

29
Mielofibrose Primária
Tratamento baseado em grupos de risco ( Cervantes) Idade < 55 anos Baixo risco: tratamento de suporte; Alto risco: TMO alogênico: SG 38-58%; RIC: possível em > 55 anos, SG 80-84%. Idade > 55 anos Tratamento de suporte ( considerar RIC).
 Idade < 55 anos. Baixo risco: tratamento de suporte; Alto risco: TMO alogênico: SG 38-58%; RIC: possível em > 55 anos, SG 80-84%. Idade > 55 anos. Tratamento de suporte ( considerar RIC).")
30
Mielofibrose Primária
Tratamento de suporte Anemia: - Transfusão - Andrógenos (em pacientes sem alterações citogenéticas ou citopenia severa) - EPO altas doses de U/Kg/semana (em pacientes com EPO sérica < 123 e baixa necessidade transfusional) - Talidomida + prednisona
 - EPO altas doses de U/Kg/semana (em pacientes com EPO sérica < 123 e baixa necessidade transfusional) - Talidomida + prednisona.")
31
Mielofibrose Primária
Tratamento de suporte Quimioterapia Indicada para pacientes com sintomas constitucionais, hiperproliferação e esplenomegalia sintomática progressiva Maior experiência com Hidroxiuréia mg/Kg/dia, INF 5mU/dia pode ser utilizado em pacientes jovens (50% resposta)
")
32
Mielofibrose Primária
Tratamento de suporte Esplenectomia Normalmente reservada para pacientes com esplenomegalia sintomática ou citopenia com alta necessidade transfusional; Aumenta o risco de transformação para LMA (27% para 55%) ( em estudo europeu); Não aumenta sobrevida global.
 ( em estudo europeu); Não aumenta sobrevida global.")
33
Mielofibrose Primária
Tratamento de suporte Irradiação esplênica - Para pacientes não canditados à esplenectomia - Baixas doses (2.8 a 24 Gy) para evitar citopenia grave - Reverte dor e esplenomegalia em 93% - Citopenia significativa em 43%, citopenia grave em %, 13% de mortalidade por sepse ou sangramento - Duração mediana da resposta de 6 meses (1 a 41) - Não há evidência de aumento de risco de LMA
 para evitar citopenia grave. - Reverte dor e esplenomegalia em 93% - Citopenia significativa em 43%, citopenia grave em 26%, 13% de mortalidade por sepse ou sangramento. - Duração mediana da resposta de 6 meses (1 a 41) - Não há evidência de aumento de risco de LMA.")
34
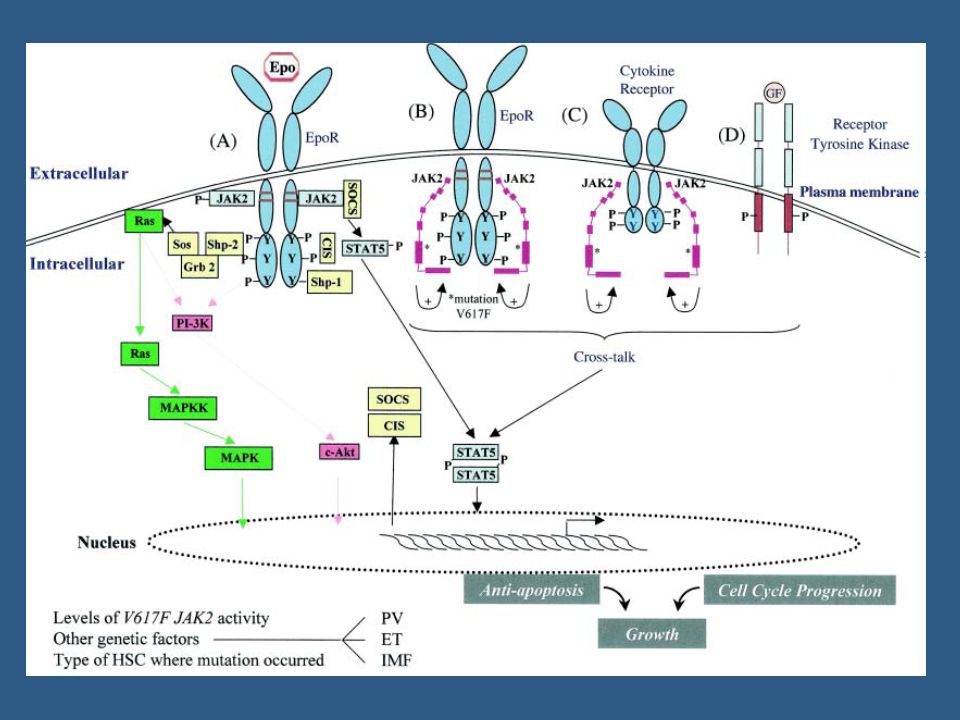
JAK 2 JAK 2: tirosino kinase citoplasmática → ativada por receptores de citocinas→ fosforila resíduos tirosina de receptores que se ligam a proteínas STAT (que ativam a transcrição gênica); JAK2V617F: mutação somática no exon 14 do gene JAK2→ troca de uma valina por uma feninalanina no códon 617 →↑atividade kinase do JAK2
; JAK2V617F: mutação somática no exon 14 do gene JAK2→ troca de uma valina por uma feninalanina no códon 617 →↑atividade kinase do JAK2.")
36
JAK 2 Técnicas para detecção da mutação: Quantitativo ou não;
AS-PCR (muito sensível); Enzima de restrição (necessita clone >5%); Sequenciamento (necessita clone>20%); Homozigotos: recombinação mitótica Homozigozidade: pode ser definida se usado método quantitativo→quando >50% tem mutação.
; Enzima de restrição (necessita clone >5%); Sequenciamento (necessita clone>20%); Homozigotos: recombinação mitótica. Homozigozidade: pode ser definida se usado método quantitativo→quando >50% tem mutação.")
37
JAK2 PV: até 99%, 1/3 homozigoto; TE: ~50%, raro homozigoto;
MF: ~50%, 1/3 homozigoto. PV: JAK 2V617F - ? Pode ter mutação no exon 12.

38
JAK 2 Impacto prognóstico:
2 estudos: não mostraram valor prognóstico em TE (150 pacientes) ou em MF (117 pacientes); Estudo europeu: ↑ mortalidade em MF; TE: ↑ risco de transformação para PV; TE: ↑ risco de trombose venosa?
 ou em MF (117 pacientes); Estudo europeu: ↑ mortalidade em MF; TE: ↑ risco de transformação para PV; TE: ↑ risco de trombose venosa")
Apresentações semelhantes
 – Lápides 1, 2, 3» «nomes gravados, 21 de Agosto de 2008» «Ultramar.TerraWeb»>")










. Nenhuns direitos reservados, excepto para fins comerciais. Por favor, não coloque.>")







