Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Doença de Hirschsprung
Monitoras: Niara Oliveira e Ângela Piccoli Ziegler Disciplina de Genética Humana FFFCMPA

2
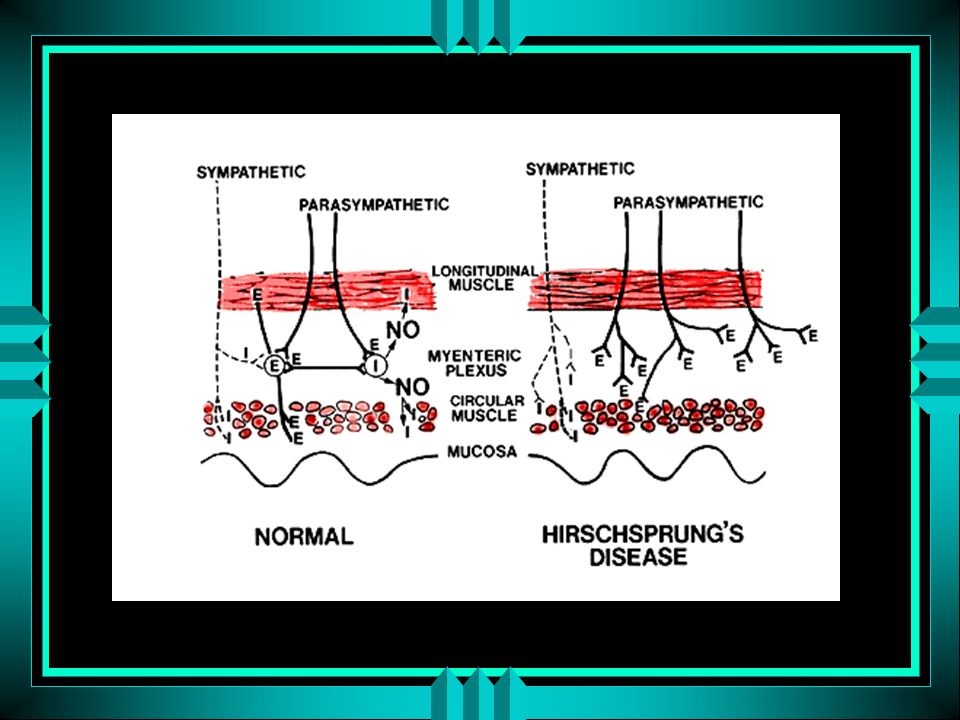
Histórico 1888: Harald Hirschsprung descreveu 2 garotos com constipação crônica , disten-são abdominal e megacolon 1940: ausência de células ganglionares intramurais dos plexos mioentéricos e submucosos 1948: Swenson e Bell desenvolveram procedimento cirúrgico – transmissão familiar 1973: Bolande propôs o termo neurocristo-patia

3
Definição Malformação congênita do intestino caracterizada pela ausência de células gangliônicas intrínsecas parassimpáticas nos plexos submucoso e mioentérico

4
Epidemiologia incidência: 1:5000 nascidos vivos raça
homens 4:1mulher (forma clássica) 4% prematuros aganglionose colônica total: feminino igual a masculino;
 4% prematuros. aganglionose colônica total: feminino igual a masculino;")
5
Epidemiologia Ocorre como uma característica isolada em 70% dos casos
Associação com anormalidade cromossô-mica em 12% dos casos Associação com anormalidades congênitas adicionais em 18% dos casos (algumas síndromes monogênicas )
")
6
Patologia Parada prematura da migração craniocaudal das células vagais da crista neurais no intestino grosso entre a 5ª a 12ª semana de gestação para formar o sistema nervoso entérico (neurocristopatia)
")
7
Patologia 5a semana: precursores de neuroblastos no intestino primitivo anterior 8a semana: colon transverso 12a semana: reto após a migração caudal dos neuroblastos, ocorrem a distribuição e a migração dos mesmos para camadas mais superficiais e mais profundas da parede intestinal, seguidas de maturação dos neuroblastos para célula ganglionares

8
Patologia fatores imunológicos: destruição de células ganglionares (?)
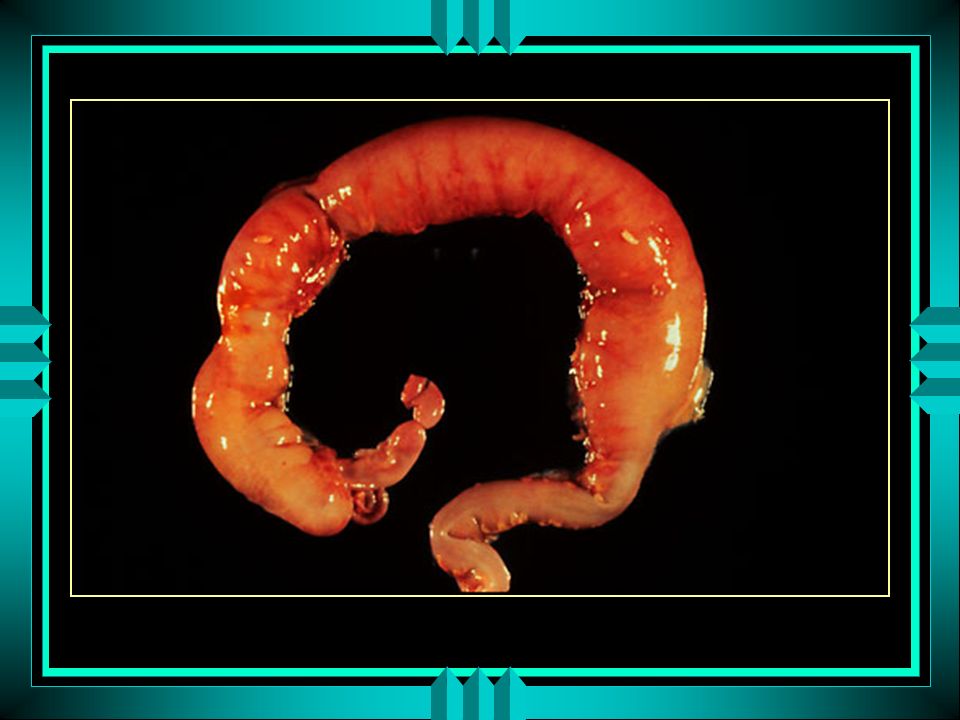
ausência de células ganglionares nos feixes intermuscular (Auerbach) e submucosas (Meissner) do intestino zona aganglionar é aperistálica, espástica, contrai-se em bloco
 e submucosas (Meissner) do intestino. zona aganglionar é aperistálica, espástica, contrai-se em bloco.")
9
Patologia segmentos proximais ganglionares se dilatam
hipertrofia muscular e parede espessada vasos da parede se dilatam esfíncter interno do reto também tem sua inervação alterada entre as zonas espástica e dilatada, há uma zona de transição hipoganglionar (graus variáveis de hipertrofia de fibras nervosas; zona doente)
")
11
Classificação Forma curta: segmento agangliônico não se estende além do sigmóide (80%) Forma longa: segmento agangliônico se estende proximalmente ao sigmóide (20%) Limite inferior: esfíncter anal externo
 Limite inferior: esfíncter anal externo.")
12
Variantes: Aganglionose colônica total (3-8%) Aganglionose intestinal total Forma ultra-curta (reto distal) Aganglionose setorial (?): porção agangliônica está acima de um segmento distal normal.
: porção agangliônica está acima de um segmento distal normal.")
13
Características Clínicas
Obstrução intestinal com as seguintes características: Falência na passagem do mecônio em 48h Distensão abdominal que alivia com estimulação retal ou enema Vômitos Enterocolite neonatal Desnutrição crônica

14
Características Clínicas
A intensidade da sintomatologia depende mais da maior quantidade de fibras colinérgicas no reto (maior espasticidade), do que da extensão da zona aganglionar
, do que da extensão da zona aganglionar.")
16
Diagnóstico 50% das crianças tem o diagnóstico feito no período neonatal Radiografia simples de abdome Enema opaco Manometria anorretal Biópsia da parede retal Exames histológicos: coloração HE; pesquisa da atividade da acetilcolinesterase

17
Alguns pacientes são diagnosticados mais tardiamente na infância ou até na idade adulta com constipação severa, distensão abdominal crônica, vômitos Raramente se apresenta por perfuração inexplicada do ceco ou do apêndice

18
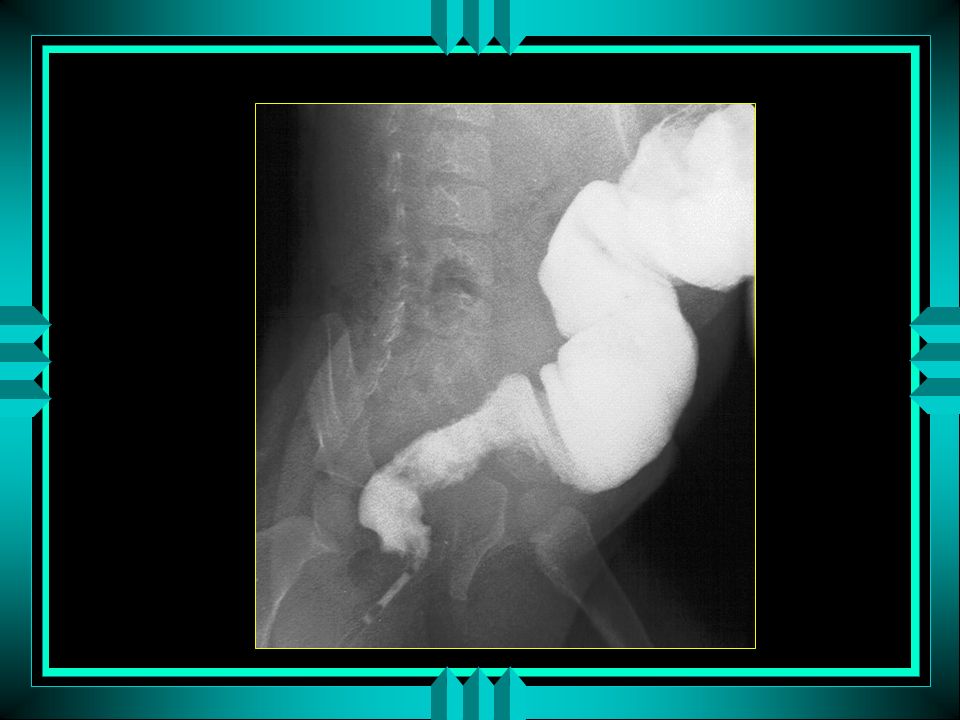
Radiologia Distensão do intestino delgado e colon proximal com reto vazio Algumas vezes é possível identificar zona retal espástica Não dá diagnóstico de certeza

19
Enema Opaco Bário (não tem efeito laxativo)
Presença de zona espástica ao nível do reto e dilatação dos segmentos intestinais acima Extensão do segmento agangliônico Mais significativo quanto maior a idade e a espasticidade da zona acometida

20
Enema Opaco Não é sensível nem específico no período neonatal
Sem preparo prévio Obter um radiografia de retardo de 24 horas

22
MANOMETRIA ANORRETAL não é útil: RN com IG 39 semanas; 12 dias de vida útil como método de exclusão um crianças com constipação intestinal método de triagem nos casos de megarreto, criança com colostomia prévia e nos casos de diagnóstico duvidoso.

23
EXAMES HISTOLÓGICOS coloração pela hematoxilina – eosina (ausência de células ganglionares e existência de troncos hipertrofiados) pesquisa da atividade de acetilcolinesterase (aumentada no HSCR)
")
24
Biopsia da Parede Retal
ausência de células ganglionares nos plexos hipertrofia das camadas musculares; fibras nervosas pré-ganglionares hipertrofiadas (provenientes da cadeia parassimpática paravertebral

25
Diagnóstico diferencial
Íleo meconial ( fibrose cística) Malformação intestinal: atresia intestinal baixa ou colônica, mal rotação ou duplicação intestinal, etc Anormalidade do SNE: síndromes de pseudo – obstrução intestinal crônica Obstrução intestinal funcional resultante de infecção materna, intoxicação materna, hipotireoidismo congênito
 Malformação intestinal: atresia intestinal baixa ou colônica, mal rotação ou duplicação intestinal, etc. Anormalidade do SNE: síndromes de pseudo – obstrução intestinal crônica. Obstrução intestinal funcional resultante de infecção materna, intoxicação materna, hipotireoidismo congênito.")
26
Clinicamente a doença de HSCR pode ser dividida em:
Etiologia Clinicamente a doença de HSCR pode ser dividida em: Doença de HSCR Sindrômica Doença de HSCR Isolada

27
Doença de HSCR Sindrômica
Síndrome de Lesch-Nyhan Displasia Mesomélica Miscelânea HSCR com anormalidades de membros Síndrome de Kaplan Goldberg-Shprintzen Síndrome de Clayton-Smith HSCR mandatória Distrofia muscular congênita de Fukuyama Síndrome de Riley-Day HSCR raramente associada Síndrome da Hipoventilação Central Congênita Síndrome de Smith-lemli-Optiz MEN 2 Síndrome de McKusick-Kauffman Síndrome Waadenburg HSCR ocasional Neurocristopatias

28
Alterações Cromossômicas
Síndrome de Down A trissomia livre do 21 é a alteração citogenética mais comumente associada à Doença de HSCR (2 a 10% dos casos de HSCR). Nesses casos, a prevalência de S-HSCR e do sexo masculino é ainda maior que nos casos não-sindrômicos.
. Nesses casos, a prevalência de S-HSCR e do sexo masculino é ainda maior que nos casos não-sindrômicos.")
29
Alterações Cromossômicas
Síndrome de Down A superexpressão dos genes do cromossomo 21 como predisposição ao desenvolvimento da doença de HSCR é uma das hipóteses. (gene suscetível mapeado na região 21q22)
")
30
Alterações Cromossômicas
Microdeleções Algumas deleções intersticiais associadas a HSCR têm sido importantes para a identificação de genes predisponentes: Deleção intersticial 10q11.2 em pacientes com L-HSCR e aganglionose total (que levou à identificação e mapeamento do primeiro gene para doença de HSCR);
;")
31
Alterações Cromossômicas
Deleção intersticial 13q em pacientes com S-HSCR (que levou ao mapeamento de um segundo gene EDNRB); Síndrome da deleção intersticial 2q22-23 em pacientes com HSCR (que levou à identificação do gene SIP1)
; Síndrome da deleção intersticial 2q22-23 em pacientes com HSCR (que levou à identificação do gene SIP1)")
32
Alterações Cromossômicas
Outras anormalidades cromossômicas raras já foram descritas em associação com HSCR: Síndrome de DiGeorge, mosaico para trissomia do 8, Síndrome XXY; duplicação parcial do cromossomo 2q, tetrassomia 9p e deleção 20p.

33
Alterações Cromossômicas
N0. Casos Gene Características Clínicas Cromossomo 4 ? Síndrome dos Olhos de Gato Tri 22pter-q11 MCA/RM Del 17q21 3 SIP1 Microcefalia, RM, epilepsia, dismorfias faciais e HSCR Del 2q22 7 EDNRB RM, retardo crescimento, dismorfias e S-HSCR Del 13q22 2 RET RM e L-HSCR Del 10q11 2 a 10% casos HSCR Síndrome de Down e S-HSCR Tri 21

34
Doença de HSCR Sindrômica
Na doença de HSCR Sindrômica o padrão de herança obedece ao padrão de herança característico de cada síndrome.

35
A doença de HSCR isolada é considerada uma Doença de Herança Complexa
Pode se apresentar com padrão de herança mendeliana monogênica ou com padrão de herança multifatorial.

36
of a complex inheritance
Hirschsprung disease as genetic model of a complex inheritance Genetic Heterogeneity Synergistic Mutations in anyone of several genes are sufficient for phenotype expression of HSCR The cumulative effect of mutations in multiple genes contributes to the phenotype in one individual The interaction of both mechanisms determines all disease phenotypes in a HSCR population

37
Doença de HSCR Isolada Uma mutação em apenas um dos genes predisponentes pode causar o fenótipo da doença de HSCR – Herança Monogênica Mutações em diversos genes podem determinar em conjunto o fenótipo de HSCR – Herança Multifatorial

38
Doença de HSCR Isolada Independentemente, o principal gene envolvido no desenvolvimento da Doença de HSCR é o RET.

39
Alterações Moleculares
RET (“rearranged during transfection”) Estima-se que mutações no RET sejam responsáveis por mais de 50% dos casos de HSCR isolada.
 Estima-se que mutações no RET sejam responsáveis por mais de 50% dos casos de HSCR isolada.")
40
Alterações Moleculares
Outros genes envolvidos no desenvolvimento da doença de HSCR incluem: GDNF (5p13) EDNRB (13q22) NTN (19p13) SOX10 (22q13)
 EDNRB (13q22) NTN (19p13) SOX10 (22q13)")
41
Características do gene RET
Definição: É um proto-oncogene que codifica uma glicoproteína transmembrana relacionada à família dos receptores tirosina-kinase. Localização: 10q11.2 Tamanho: 60 Kb/21 éxons (O íntron 1 corresponde a 24Kb, ficando o restante do gene (éxons 2 a 20) dispostos em apenas 31kb).
 dispostos em apenas 31kb).")
42
Características do gene RET
Proteína: Codifica uma proteína transmembrana tirosina-kinase, similar às demais proteínas da família de receptores tirosina-kinase, exceto pela presença de um domínio caderina-like (responsável pela adesão célula-célula) e uma região rica em cisteína na porção extracelular.
 e uma região rica em cisteína na porção extracelular.")
43
Características do gene RET
Isoformas: Devido ao mecanismo de splicing alternativo do RNAm, a proteína RET é expressa em duas principais isoformas, RET 43 e RET 51. A RET 51 é a maior das isoformas, com 1114 aminoácidos. É co-expressa com a RET 9 na maioria dos tecidos.

44
Mecanismo de Ativação do RET
‘Ménage à trois’ O RET se associa ao GFR-1 (“GDNF family receptors”) e forma receptores para a família de ligantes GDNF (“glial-cell-line-derived neurotrophic factor”). O GDNF faz parte da família dos GFLs (“glial-cell-line-derived neurotrophic factor ligands”), que também inclui NRTN (“neurturin”), ARTN (“artemin”) e PSPN (“persephin”).
 e forma receptores para a família de ligantes GDNF ( glial-cell-line-derived neurotrophic factor ). O GDNF faz parte da família dos GFLs ( glial-cell-line-derived neurotrophic factor ligands ), que também inclui NRTN ( neurturin ), ARTN ( artemin ) e PSPN ( persephin ).")
46
RET-Receptor complex GDNF NTN GFR1 GFR2 Extracellular Intracellular
TK2 TK1 CD CYS CD GFR1 GFR2 Extracellular CYS Cell membrane TK1 Intracellular RET TK2 Gentilmente cedido por Alexandre Serra

47
Mecanismo de Ativação do RET
A molécula dimérica de GDNF se liga a dois monômeros de GFR-1 e esse complexo se liga ao RET, promovendo sua homodimerização e ativação.

48
Mecanismo de Ativação do RET
O RET não é capaz de se ligar ao GDNF na ausência do GFR-1. No entanto, a afinidade desses dois compostos entre si não é modificada pela presença ou não do RET. Por isso, mutações que afetam apenas o GDNF também podem causar HSCR (causa rara, menos de 5% dos casos).
.")
49
Mecanismo de Ativação do RET
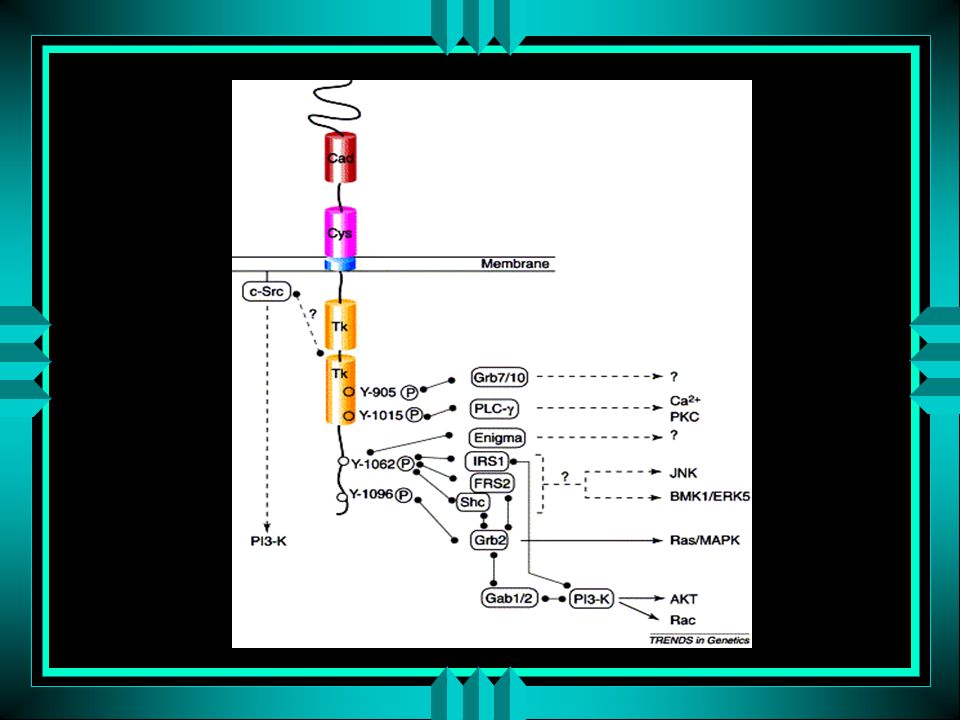
A ativação do RET promove a autofosforilação dos resíduos de tirosina-kinase na cauda citoplasmática, criando locais de sinalização para múltiplos segundos-mensageiros (PI3K, JNK, PLC, Ras-MAP kinase).
.")
51
Papel do RET no desenvolvimento do SNP
O RET é expresso em uma população de células da crista neural que migram do neuroepitélio cerebral e colonizam todo o tubo digestivo, formam o gânglio simpático cervical superior e dão origem às células C da tireóide.

52
Papel do RET no desenvolvimento do SNP
Células RET + são detectadas durante a sua migração no mesênquima intestinal e subseqüentemente são observadas nos plexos nervosos (Plexos de Auerbach e Meissner) que formam o SNE.
 que formam o SNE.")
53
Papel do RET no desenvolvimento do SNP
As células que migram da crista neural para o tubo digestivo co-expressam RET e GFR-1 e dependem da interação com o GDNF durante a migração ou no local de gangliogênese para que sejam funcionais.

54
Papel do RET no desenvolvimento do SNP
O GDNF é expresso em todo mesênquima embrionário gastrintestinal – é um potente quimiotrófico, mitogênico e fator trófico de diferenciação para precursores entéricos derivados da crista neural. O GDNF promove a proliferação e a sobrevivência dos progenitores do SNE cultivados in vitro.

56
O RET também é expresso em tumores derivados de células da crista neural, como neuroblastomas, carcinomas medulares de tireóide e feocromocitomas – o que sugere a participação do gene no controle da proliferação, diferenciação, migração e funcionalidade das células da provenientes da crista neural!

57
RET e HSCR Fisiopatologia
Mutações no RET são responsáveis por mais de 50% dos casos familiares de HSCR isolada. As mutações distribuem-se por toda a extensão da seqüência codificadora – não há um hot spot bem estabelecido (umas das poucas coisas que se sabe com certeza!) .
 .")
58
Os tipos de mutações que podem ocorrer incluem:
Mutações do RET Os tipos de mutações que podem ocorrer incluem: Deleções e inserções Mutações frameshift Mutações missense e nonsense Alterações de splicing

59
Mutações do RET As conseqüências funcionais das mutações estão relacionadas à sua posição na seqüência codificadora e são classificadas em 4 grupos: Mutações Classe I: localizadas no domínio extracitoplasmático, alteram a maturação do RET e inviabilizam o envio do sinal através da membrana plasmática

60
Mutações do RET Mutações Classe II: causam substituição de um dos 4 resíduos extracitoplasmáticos de cisteína por um aa diferente, provendo uma alteração constitutiva na proteína, que diminui sua presença na superfície celular

61
Mutações do RET Mutações Classe III: afetam o domínio tirosina-kinase, diminuindo sua função catalítica (diminuem sinalização) Mutações Classe IV: não diminuem sua atividade tirosina-kinase, mas interferem especificamente na transmissão do sinal para os segundos-mensageiros

62
NH2 COOH extracellular intracellular 1. Impairment of RET transport to
Gentilmente cedido por Alexandre Serra 1. Impairment of RET transport to the cell membrane 2. Impairment of RET transport to the cell membrane and covalent dimerization 3. Complete or partial loss of RET kinase activity 4. Disruption of the binding of transduction proteins on Tyr 1062 TM NH2 TK2 TK1 COOH CD CYS 5' Exon Skipping 3' alternative Splicing extracellular intracellular

63
Fisiopatologia As mutações do RET identificadas em pacientes com HSCR são todas em heterozigose, indicando que uma única cópia do gene normal é insuficiente para o desenvolvimento adequado do SNE. Isso pode ser explicado em termos de HAPLOINSUFICIÊNCIA.

64
Correlação Fenótipo x Genótipo
Cada uma dessas mutações tem efeito específico, dependendo do tecido onde o RET está sendo expresso. Podem resultar tanto em proliferação celular descontrolada (ganho de função) quanto em indução de apoptose (perda de função).
 quanto em indução de apoptose (perda de função).")
65
RET proto-oncogene Neurocristopathy Pluripotential precursor cells
Gentilmente cedido por Alexandre Serra RET proto-oncogene Activation Inactivation Neurocristopathy - Multiple Endocrine Neoplasia type 2 - Sporadic MTC - Hirschsprung disease - Ondine‘s curse ? Embryo- genesis Pluripotential precursor cells of the neural crest

66
Correlação Fenótipo x Genótipo
É muito pobre a correlação fenótipo X genótipo na doença de HSCR! Mutações isoladas no gene RET são responsáveis pelo fenótipo, no entanto, mutações isoladas em outros genes predisponentes também podem determinar o desenvolvimento da doença.

67
Tratamento Depende da extensão de cólon comprometida:
Colectomias parciais com anastomose primária Colectomia total com construção de bolsa ileal (J-Pouch) Derivação do trânsito intestinal (Duhamel)
 Derivação do trânsito intestinal (Duhamel)")
68
Aconselhamento Genético
Nos casos de Doença de HSCR monogênica (mutação no RET), o padrão de herança é autossômico dominante com penetrância incompleta, expressividade variável e sexo-dependente. Mutações em outros genes também podem seguir padrão de herança mendeliana.
, o padrão de herança é autossômico dominante com penetrância incompleta, expressividade variável e sexo-dependente. Mutações em outros genes também podem seguir padrão de herança mendeliana.")
69
Aconselhamento Genético
Nos casos isolados de HSCR, um adequado risco relativo para a prole do probando pode ser calculado levando em consideração o sexo e tamanho do segmento acometido no probando e o sexo da prole (Paradoxo de Carter).
.")
70
Aconselhamento Genético
5% / 3% 33% / 9% Probando feminino 5% / 1% 17% / 13% Probando masculino Risco de Recorrência (irmãos homem:mulher) 17:4 52:40 Penetrância (%) (homem:mulher) Multifatorial Dominante Modelo de Herança 5,5 1,75 Razão por sexo (H:M) 81 19 % de probandos S-HSCR L-HSCR
 17:4. 52:40. Penetrância (%) (homem:mulher) Multifatorial. Dominante. Modelo de Herança. 5,5. 1,75. Razão por sexo (H:M) % de probandos. S-HSCR. L-HSCR.")
71
Aconselhamento Genético
O risco de recorrência global da doença de HSCR na irmandade de um indivíduo afetado é de 4%. Lembrar: Pobre correlação fenótipo X genótipo!!

72
Aconselhamento Genético
Não está indicado screening de mutações para doença de HSCR, exceto a testagem para mutações no éxon 10 e 11 do gene RET, que são locais de mutações predisponentes ao desenvolvimento da Síndrome MEN2A.

73
Agradecimentos especiais à Prof. Carla Graziadio e à Prof. Beth
“The dream is over!” Agradecimentos especiais à Prof. Carla Graziadio e à Prof. Beth

Apresentações semelhantes







 Daniela Augustin Silveira.>")













