Carregar apresentação
A apresentação está carregando. Por favor, espere

1
OSTEOGENESIS IMPERFECTA
Denise Castro Marília Ertel Rachid Karam Rafael Bonfá FabianeVargas Cláudio Alexandre Fundação Faculdade Federal de Ciências Médicas de Porto Alegre

2
INTRODUÇÃO Osteogenesis Imperfecta (OI) é uma desordem hereditária do tecido conjuntivo que afeta a estrutura e a função dos tecidos que contêm colágeno. Defeitos moleculares do colágeno tipo I as principais causas desta doença Amplo espectro de características clínicas
 é uma desordem hereditária do tecido conjuntivo que afeta a estrutura e a função dos tecidos que contêm colágeno. Defeitos moleculares do colágeno tipo I as principais causas desta doença. Amplo espectro de características clínicas.")
3
INTRODUÇÃO Inteligência é normal
É caracterizada por diminuição da massa óssea, produzindo ossos frágeis Freqüentemente associada com: esclera azul anormalidades dentária (Dentinogenesis Imperfecta) surdez pré-senil história familiar positiva Inteligência é normal
 surdez pré-senil. história familiar positiva. Inteligência é normal.")
4
HISTÓRICO 1000 a.C: Múmia egípcia Século IX: “Ivan, o sem ossos”
1715: P. Amand Primeira descrição médica 1831: Edmund Axmann Primeira menção a esclera azul 1849: Willem Vrolik Denominação da OI 1979: David Sillence Classificação

5
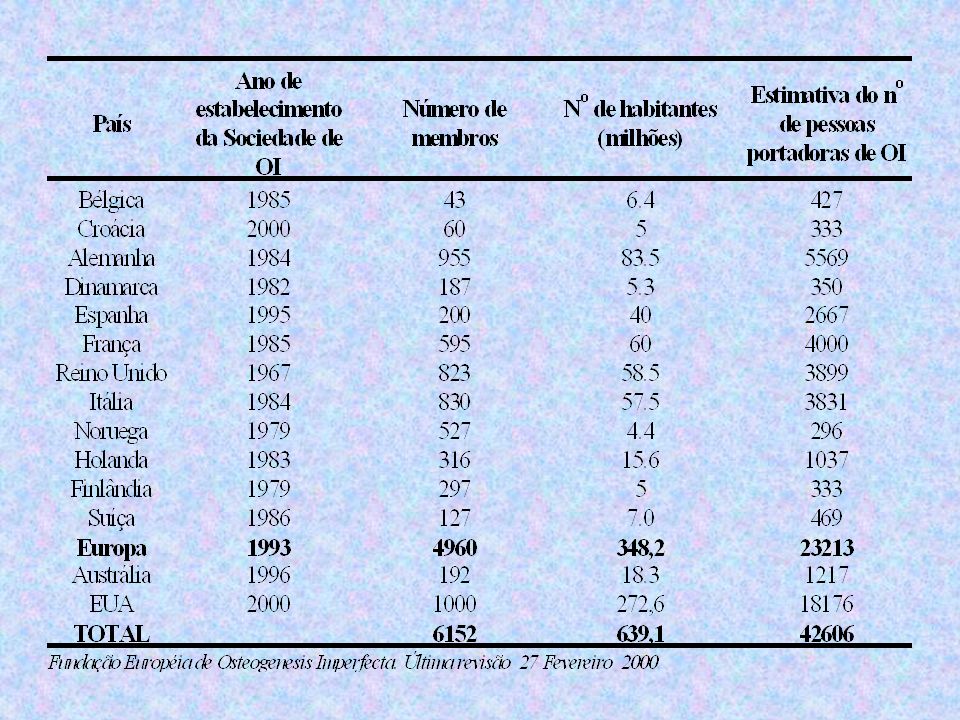
EPIDEMIOLOGIA Incidência: 1:21.000 a 1:50.000 nascidos
OI-I é a mais freqüente (1:30.000) OI-II 1:60.000 Na África, a forma mais freqüente é OI-III com herança autossômica recessiva
 OI-II 1: Na África, a forma mais freqüente é OI-III com herança autossômica recessiva.")
7
CLASSIFICAÇÃO Classificação de Sillence, 1979 define 4 tipos de OI
OI tipo I OI tipo II OI tipo III OI tipo IV Têm-se proposto um novo tipo de OI (OI tipo V) As exceções
 As exceções.")
8
OI tipo I Forma mais leve e mais comum Caracterizada por:
esclera azul permanente surdez prematura fragilidade óssea leve a moderada subdividida em: A) dentes normais B) Dentinogenesis Imperfecta
 dentes normais. B) Dentinogenesis Imperfecta.")
9
OI tipo II Forma mais severa Letal perinatal Caracterizada por:
fraturas pré-natais membros pouco desenvolvidos e curvos extrema fragilidade óssea

10
OI tipo III Forma severa não-letal Caracterizada por:
algumas fraturas ao nascimento deformidade progressiva severa nos membros e coluna vertebral esclerótica azulada tende a diminuir com a idade

11
OI tipo IV Moderadamente severa Características similares à OI-I
Esclerótica azulada diminui progressivamente Também é subdividida em: A) dentes normais B) Dentinogenesis Imperfecta
 dentes normais. B) Dentinogenesis Imperfecta.")
12
CLASSIFICAÇÃO

13
COLÁGENO Proteína mais comum no mundo animal
Colágeno tipo I é o mais abundante Principal proteína estrutural da matriz extracelular dos ossos, pele e tendões Forma 90% da matriz extracelular óssea

14
COLÁGENO ESTRUTURA Duas cadeias -1 e uma cadeia -2
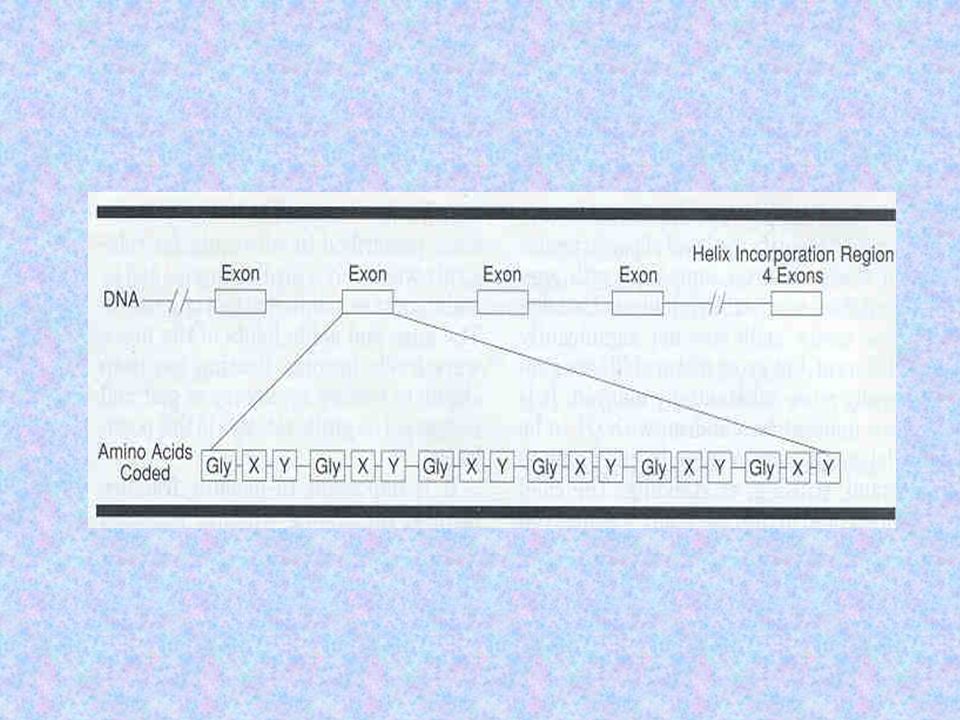
Cada cadeia contém: pró-peptídios C-terminal e N-terminal domínio central composto de 338 repetições de Gly-X-Y, onde o X e o Y representam qualquer outro aminoácido que não a glicina

15
COLÁGENO ESTRUTURA Por ser o menor aminoácido, a glicina é o único capaz de ocupar a posição axial da tripla hélice Qualquer alteração na glicina acarretará desorganização da estrutura do colágeno

17
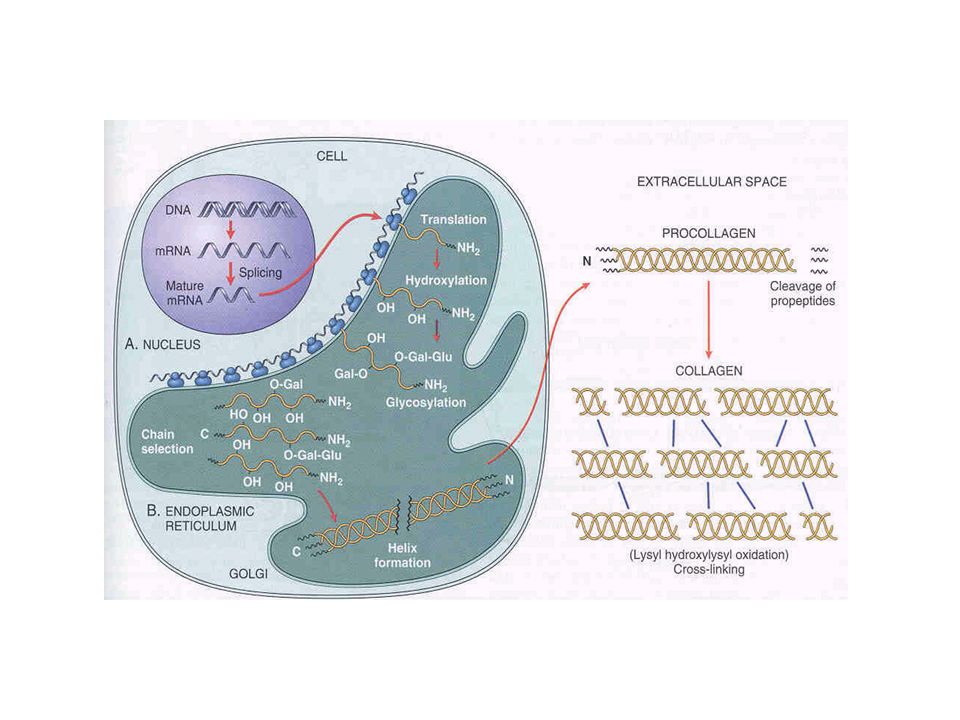
COLÁGENO SÍNTESE Processo intracelular complexo para entrelaçamento das três pró-cadeias Processo extracelular de agrupamento de colágeno em microfibrilas

18
COLÁGENO mRNA maduro é obtido pelo splicing do mRNA
mRNA maduro + Ribosomos: síntese das pró-cadeias Terminais livres traduzidos movem-se para o interior do RE-rugoso

19
COLÁGENO Peptídeos hidrofóbicos sinalizadores no terminal amino são clivados: Sinal para o início das modificações pós-traducionais Resíduos de prolinahidroxiprolina, pela prolina hidroxilase Resíduos de lisinahidroxilisina, pela lisina hidroxilase Resíduos de hidroxilisina são glicosilados com galactose associada ou não a glicose

20
COLÁGENO Quando os últimos aminoácidos são incorporados, as pró-cadeias são liberadas dentro das cisternas do RE-rugoso 2 pró-cadeias -1 e 1 pró-cadeia -2 unem-se por seus pró-peptídeos C-terminais A estrutura é mantida por pontes dissulfídricas

21
COLÁGENO Modificações pós-traducionais continuam até que cada cadeia adquira por volta de 100 resíduos de hidroxiprolina Seqüências Gly-X-Y ricas em hidroxiprolina na posição C-terminal entrelaçam-se A região C-terminal torna-se núcleo para a auto-montagem da tripla hélice

22
COLÁGENO A conformação de tripla hélice em uma seqüência Gly-X-Y induz o entrelaçamento da próxima seqüência O resultado é a propagação como um zíper no sentido C-terminal para N-terminal A proteína passa para o Golgi onde é secretada

23
COLÁGENO Após a secreção, pró-colágeno é processado a colágeno por clivagem dos pró-peptídios N-terminais e C-terminais por proteases Por ser menos solúvel que o pró-colágeno, o colágeno se agrega formando as fibrilas.

24
COLÁGENO Oxidações dos resíduos amino da lisina e hidroxilisina pela lisina-oxidase possibilitam ligações covalentes cruzadas entre as cadeias de uma mesma molécula e de moléculas adjacentes conferindo a força tensil do colágeno

26
COLÁGENO GENES Gene da pró-cadeia -1 (COL1A1): banda 17q21.31-q22
: banda 17q21.31-q22")
28
PATOLOGIA MOLECULAR

29
MATRIZ EXTRACELULAR Os animais secretam uma complexa rede de proteínas e carboidratos, a MEC, criando um ambiente especial entre as células. A MEC é fundamental para ligar as células em tecidos. A MEC também propicia um meio por onde as células podem mover-se, particularmente durante os estágios iniciai da diferenciação.

30
Defeitos entre as conexões célula-MEC levam a malformações congênitas, como ocorre na OI

31
1. A glicina é o menor AA e, portanto, o único resíduo compacto o bastante para adaptar-se perfeitamente à posição axial na formação da tripla hélice

32
A substituição do resíduo de glicina por um AA maior é altamente desorganizador da estrutura helicoidal da molécula de colágeno tipo I GLICINA CISTEÍNA

33
2. A montagem das cadeias individuais na tripla hélice começa na extremidade Carboxi-terminal e move-se em direção à extremidade Amino-terminal

34
Portanto, mutações proximais à região C-terminal da molécula são mais desorganizadoras, porque interferem mais cedo na propagação da da hélice

35
3. As modificações pós-traducionais do pró-colágeno (hidroxilação da prolina e da lisina e glicosilação das hidroxilisinas) continuam em qualquer parte de uma cadeia não montada na tripla hélice
 continuam em qualquer parte de uma cadeia não montada na tripla hélice.")
36
Portanto, quando a montagem da hélice é diminuida por uma mutação, as seções não montadas das cadeias N-terminais ao defeito são altamente modificadas, reduzindo sua secreção e contribuindo para sua instabilidade

37
4. Pacientes com substituições da glicina dentro dos domínios das pró-cadeias 1 e 2 podem ter OI II, III ou IV. Estes pacientes apresentam anomalias qualitativas e quantitativas.

38
O que determina o fenótipo destes pacientes?

39
Hipótese: gradiente de severidade fenotípica

40
Problema: Há relatos onde a substituição da glicina por serina nas posição 352 e 415 da pró-cadeia 1 pode produzir o fenótipo de OI tipo II, III e IV.

41
Como explicar? Evento causal ambiental, como um trauma que leve a uma fratura em uma criança durante o parto Loci modificadores Relação com o sistema de Controle de Qualidade da célula

42
O Sistema de Controle de Qualidade Pós-traducional

43
A Chaperona molecular BiP , encontrada no RE, está ligada especificamente no pró-colágeno tipo I mutante Em experimentos onde inibiu-se a exportação protéica do RE, foi mostrado que pró-cadeias 1 danificadas irreversivelmente eram degradadas seletivamente dentro do próprio RE, provavelmente pelas proteases

44
4. A OI tipo I é uma anomalia quantitativa, pois não é detectável no citoplasma nenhuma proteína mutante.

45
Mecanismo: Códons de parada prematuros no gene COL1A1
Deleção de todo ou de parte do gene COL1A1 Erros no splicing do RNAm

46
As mutações variam em seu tipo e local no gene, mas o resultado funcional é o mesmo. Há uma haploinsuficiência do gene COL1A1, já que este gene não apresenta expressividade, sendo chamado de alelo nulo.

47
Resumindo: Na OI-I temos uma alteração quantitativa do colágeno tipo I
Na OI II, III e IV há alteração qualitativas e quantitativas do colágeno tipo I

48
CARACTERÍSTICAS CLÍNICAS

49
OI tipo I Fragilidade óssea sem deformidades significativas
Estatura normal Raras fraturas no período neonatal aumento das taxas de fratura na infância redução após puberdade aumento após menopausa em mulheres e após 60 anos em homens

50
OI-I; Pai e filho mostrando leve diminuição da estatura

51
Esclera azul por toda a vida
OI tipo I Esclera azul por toda a vida

52
OI tipo I Aproximadamente metade dos pacientes com 50 anos tem PERDA AUDITIVA Inicia na 2ª - 3ª década e progride com o envelhecimento Usualmente é uma perda condutiva Idosos também apresentam perda neurossensorial

53
OI tipo I Envolvimento cardiovascular
prolapso mitral insuficiência valvular aórtica estenose aórtica Essas doenças são mais freqüentes em portadores de OI do que na população em geral ???

54
OI tipo I Dentinogenesis Imperfecta algumas famílias com OI-I
entidade separada da OI com herança autossômica dominante resultante de mutação no cromossomo 4q afeta somente os dentes pior prognóstico (maiores taxas de fraturas, deformidades do esqueleto e menor estatura)
")
55
OI tipo I Raio-X osteopenia generalizada
deformidades são resultado da angulação nos sítios de fraturas prévias

56
A B

57
OI tipo II É a forma mais severa Extrema fragilidade óssea
Morte intra-uterina ou no início da infância Além das anormalidades no esqueleto está associada com alterações neuropatológicas Clínica e bioquimicamente heterogênio - 4 grupos

58
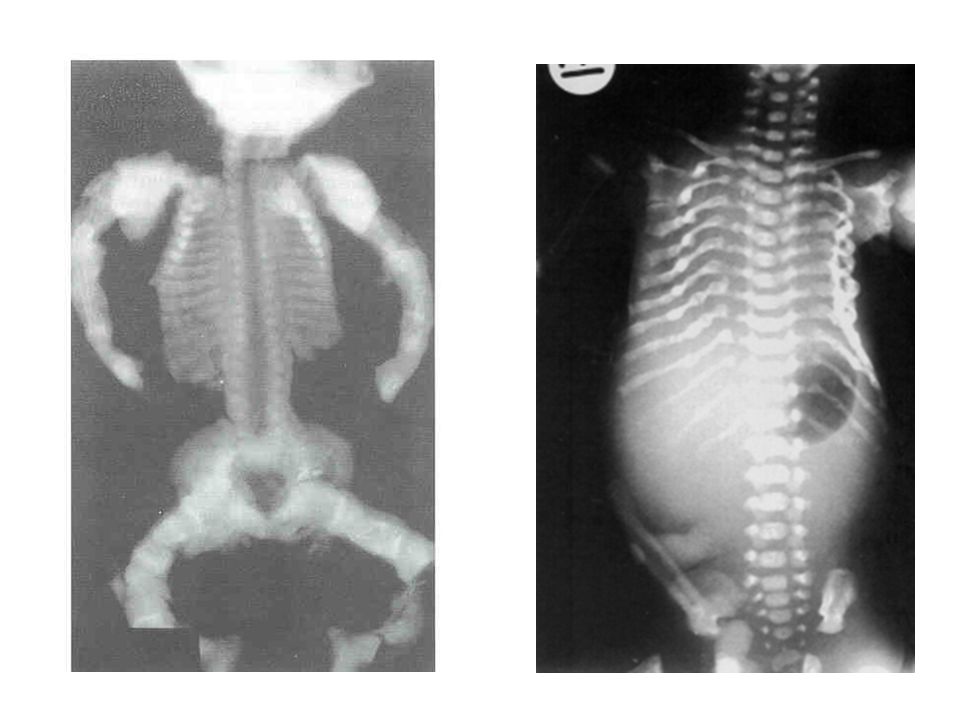
OI tipo II Peso e altura ao nascer menor que o percentil 50
Coxas fixas em rotação externa e abdução, tórax pequeno, membros curtos, angulados e curvos Escleras azuis

59
Feto portador de OI-II

60
OI tipo II Ao raio X: tórax pequeno com costelas encurtadas, espessadas, com contornos ondulados (padrão em contas) fêmur largo e retangular com margens onduladas, haste curva tíbia angulada

62
OI tipo III Manifestações neonatais de fragilidade óssea e deformidades Usualmente não-letal Apresentam múltiplas fraturas desde o nascimento deformidades esqueléticas progressivas Crescimento longitudinal pobre

63
OI tipo III Esclera azul ao nascimento com diminuição progressiva da tonalidade Dentinogenesis Imperfecta na primeira dentição Disfunção progressiva e reparo da metáfise aparência similar à pipoca

64
Criança com 6 anos mostrando severo nanismo, deformidades dos membros superiores e inferiores
Deformidades em pipoca nas regiões de metáfises do fêmur e tíbia

65
OI tipo IV Início pós-natal de fraturas Deformidade óssea leve
Perda auditiva precoce Tonalidade azulada da esclera diminui progressivamente (todos os adultos têm esclera normal) Apresentam menor estatura que portadores de OI-I
 Apresentam menor estatura que portadores de OI-I.")
66
OI tipo IV Dentinogenesis Imperfecta é observada em algumas famílias sugerindo que a OI-IV, assim como a OI-I deva ser dividida em 2 subgrupos: A (dentes normais) e B (dentina opalescente) - forma mais severa
 e B (dentina opalescente) - forma mais severa.")
67
Paciente com 2 anos, portadora de OI-IV com membros inferiores levemente curvados

68
EXPRESSIVIDADE E PENETRÂNCIA
PENETRÂNCIA define a probabilidade de um gene ter expressão fenotípica EXPRESSIVIDADE grau do efeito exercido pelo gene no fenótipo

69
EXPRESSIVIDADE E PENETRÂNCIA
Variabilidade clínica entre os tipos de OI dentro de um mesmo tipo com os mesmos defeitos moleculares é um fenômeno bem conhecido

70
EXPRESSIVIDADE E PENETRÂNCIA
Na OI-I a penetrância para fraturas é de 90% e para esclera azul de 100%. A penetrância para perda auditiva é de 43% e existe clara correlação com a idade e a progressão da perda auditiva

71
A maioria das mutações implicadas na OI são autossômicas dominantes
HERANÇA GENÉTICA A maioria das mutações implicadas na OI são autossômicas dominantes

72
A OI ilustra a complexidade genética resultante de mutações que alteram proteínas estruturais, sobretudo quando compostas de múltiplas subunidades diferentes As mutações que ocorrem em um dos alelos e que levam a mudanças estruturais na proteína reduzindo a contribuição do alelo normal são ditas mutações dominantes negativas

73
2pró-cadeias 1 : 1pró-cadeia 2
É fundamental o entendimento da relação estequiométrica do colágeno tipo I: 2pró-cadeias 1 : 1pró-cadeia 2

74
OI tipo I

75
Herança dominante Fenótipo relativamente leve, concordando com o fato de que, embora apenas metade do número de moléculas seja produzida, elas são estruturalmente normais (defeito quantitativo) Há, portanto, uma haploinsuficiência, pois um dos alelos é nulo (alelo nulo)
 Há, portanto, uma haploinsuficiência, pois um dos alelos é nulo (alelo nulo)")
76
OI tipo II, III, e IV (mutação na 1)
")
77
Se uma pró-cadeia 1 for anormal, três de cada quatro moléculas do colágeno tipo I terão pelo menos uma cadeia anormal (mutação dominante negativa) Portanto, o efeito do alelo mutante é ampliado por causa da natureza polimérica da molécula do colágeno Defeito qualitativo e quantitativo

78
OI tipo II, III e IV (mutação em 2)
")
79
Se uma pró-cadeia 2 for defeituosa, uma de duas moléculas será afetada. É também uma mutação dominante negativa Também é um defeito qualitativo e quantitativo.

80
Alguns pontos sobre OI-II:
A maioria dos bebês com OI tipo II tem uma mutação dominante nova; Em algumas famílias, mais de uma criança é afetada com a doença. Recentemente reconheceu-se mosaicismo parental que afeta a linhagem germinativa além de alguns tecidos somáticos.

81
Alguns pontos sobre OI-III:
Na maioria, a herança é dominante Existem provas de uma forma rara de herança autossômica recessiva, principalmente entre negros africanos Nestes pacientes não foi identificado defeitos nas pró-cadeias 1 e 2. Dissomia uniparental materna foi relatada em um caso

82
Alguns pontos sobre OI-IV:
É uma herança autossômica dominânte. A OI-IV é mais freqüentemente ligada a mutação no gene COL1A2.

83
Resumindo: Em doenças com mutações dominante negativas, como a OI, é melhor ter uma mutação que resulte em ausência do produto gênico (alelo nulo) do que uma mutação causadora de um produto gênico anormal
 do que uma mutação causadora de um produto gênico anormal.")
84
DIAGNÓSTICO Geralmente feito com base clínica
fraturas e deformidades ósseas esclera azul Dentinogenesis Imperfecta história familiar positiva Devem ser excluídas outras causas de fraturas patológicas

85
DIAGNÓSTICO Raio X do esqueleto Densitometria óssea
diminuição da densidade óssea fraturas calos de consolidação deformidades Densitometria óssea

86
DIAGNÓSTICO Análise molecular
Cultura de fibroblastos da derme: obtenção de colágeno para estudo molecular Eletroforese (SDS-PAGE): padrão de migração lenta Seqüenciamento: determinação da mutação
: padrão de migração lenta. Seqüenciamento: determinação da mutação.")
87
DIAGNÓSTICO PRÉ-NATAL
MÉTODOS Ultrassonografia Biópsia de vilosidades coriônicas Amniocentese Diagnóstico pré-implantação

88
DIAGNÓSTICO PRÉ-NATAL
ULTRASSONOGRAFIA 17a semana gestacional Baixa ecogeneidade de todos os ossos Movimentos severamente limitados Posicionamento anormal de membros inferiores

89
DIAGNÓSTICO PRÉ-NATAL
BIÓPSIA DE VILOSIDADES CORIÔNICAS 10a a 12a semanas gestacionais Vilos apresentam grande quantidade de colágeno para estudo Resultados em 3 a 5 dias Método de escolha para o dignóstico pré-natal

90
DIAGNÓSTICO PRÉ-NATAL
DIAGNÓSTICO PRÉ-IMPLANTAÇÃO Material de fertilização in vitro Mutação já conhecida no progenitor afetado Diagnóstico por PCR e eletroforese Diagnóstico mais precoce

91
ACONSELHAMENTO GENÉTICO
OI-I herança autossômica dominante normal + afetado 50% de recorrência ambos pais afetados 75% de recorrência

92
ACONSELHAMENTO GENÉTICO
OI-II risco de recorrência de 8,6% mutação dominante constitucional nova mosaicismo de células germinativas autossômica recessiva (rara)
")
93
ACONSELHAMENTO GENÉTICO
OI-III mosaicismo parental 7% autossômica dominante 50 a 75% autossômica recessiva 25%

94
ACONSELHAMENTO GENÉTICO
OI-IV herança autossômica dominante normal + afetado 50% de recorrência ambos pais afetados 75% de recorrência

95
TRATAMENTO MEDIDAS GERAIS
Tratamento médico tem sido largamente inefetivo em alterar o curso da doença O parto deve ser realizado da maneira menos traumática possível cesariana Menor tempo possível de imobilização evitar aparelhos gessados Iniciar fisioterapia precocemente

96
TRATAMENTO MEDIDAS GERAIS
Inserção de haste intramedular em ossos longos para prevenir e corrigir fraturas e deformidades Suporte emocional para pacientes e pais Manutenção do peso ideal obesidade implica aumento do risco para fraturas

97
TRATAMENTO MEDICAÇÕES Pamidronato Hormônio do crescimento
Estrógenos ou andrógenos Fluoreto de sódio Óxido de magnésio Calcitonina Vitamina D Pamidronato

98
TRATAMENTO PAMIDRONATO Usado e m infusões cíclicas
Melhora densidade mineral óssea Diminui as taxas de fratura Efeitos colaterais: hipersensibilidade Não se sabe dos potenciais efeitos adversos a longo prazo

99
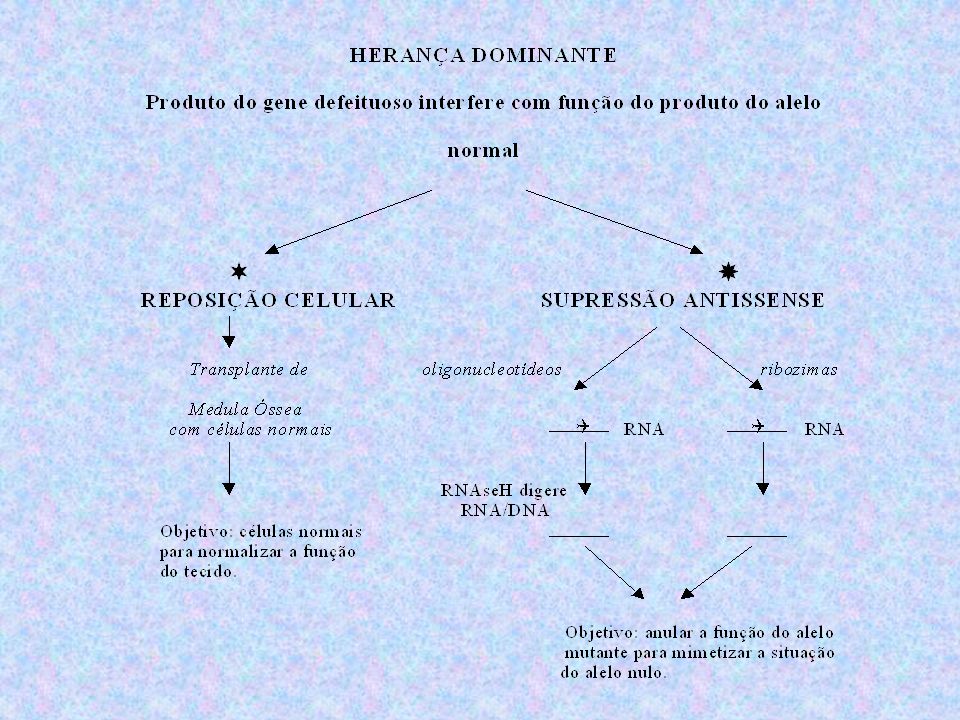
TRATAMENTO TERAPIA GÊNICA Pode ser utilizada na OI-II, III e IV
Estratégias: Reposição celular por transplante de Medula Óssea Supressão antissense Ainda está em fase de desenvolvimento

101
PROGNÓSTICO Varia de acordo com o número e severidade dos sintomas
Estatura média dos portadores de OI está abaixo da média de seus parentes de primeiro grau não afetados Expectativa de vida varia de acordo com o tipo de OI

102
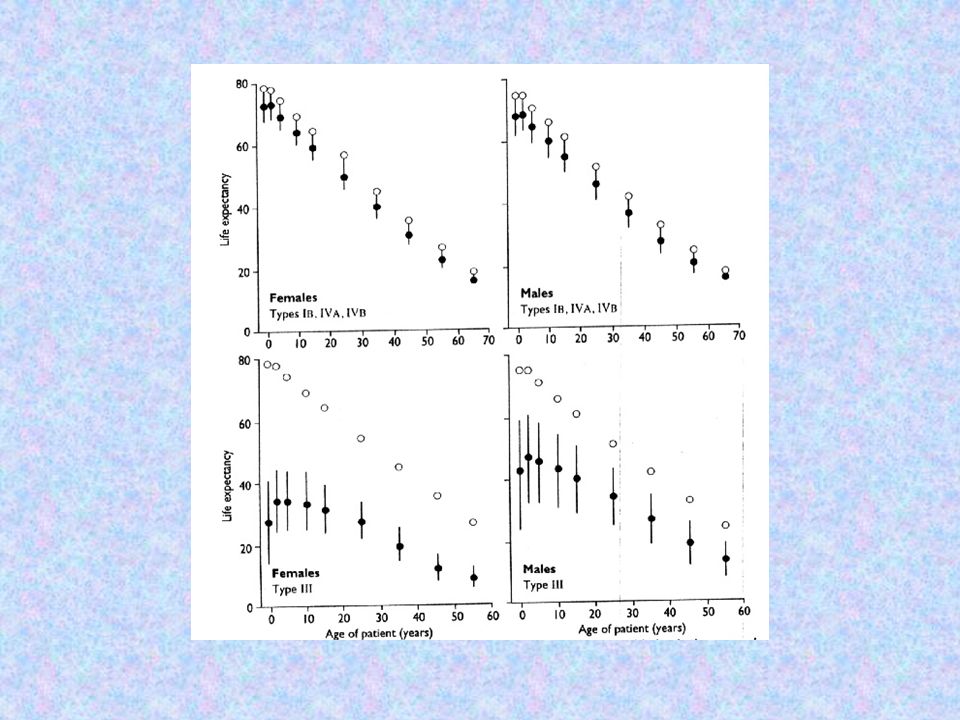
PROGNÓSTICO EXPECTATIVA DE VIDA
Tipo IA semelhante a população em geral Tipos IB e IV redução modesta Tipo II morte pré ou perinatal Tipo III redução importante

104
PROGNÓSTICO CAUSAS DE MORTE
Formas leves morte por doenças não relacionadas com a OI Formas severas morte por falência cardíaca e limitação respiratória devidas a cifoescoliose Hemorragia fragilidade vascular

Apresentações semelhantes