Carregar apresentação
A apresentação está carregando. Por favor, espere

1
BCM13042 Fundamentos de Proteínas
Aula 3 – Estratégias de Purificação e Análises de Proteínas

2
Principais componentes moleculares da bactéria E. coli
Componentes Nº de moléculas diferentes % peso total H2O Proteínas Ac. Nucléico DNA e RNA Carbohidratos Lipídeos Outros Íons Mol. Monoméricas - 500 O isolamento ou purificação de uma proteína é uma etapa que precede os estudos de suas características físico-químicas, de sua estrutura 3D e a compreensão de suas propriedades biológicas. Inicialmente, a estratégia de purificação de uma proteína é empírica, ou seja, baseada em tentativa-erro, e deve ser desenhada para cada proteína individualmente. Não há como prever quais métodos serão os mais eficientes para se purificar uma proteína pela primeira vez.

3
Métodos de Isolamento de Biomoléculas
Existe uma grande variedade de métodos visando a separação de biomoléculas. Como na maioria das vezes o que se pretende purificar é uma proteína, o grupo de moléculas com maior diversidade, as metodologias de separação de proteínas tiveram um grande desenvolvimento, com muitas opções disponíveis. Os métodos de separação de biomoléculas são agrupados em duas categorias: Métodos baseados em características físico-químicas das biomoléculas: 1. Tamanho – Massa – Densidade (ex: centrifuação, diálise, gel-filtração) 2. Carga elétrica (ex: cromatografia de troca iônica, eletroforese) 3. Solubilidade ou hidrofobicidade (ex: cromatografia em papel, fase reversa) Métodos baseados em afinidade biológica, que exploram a interação entre duas moléculas: 4. Cromatografia de afinidade (pressupõe que uma das moléculas do par que interage é um “reagente” de fácil obtenção, disponível comercialmente)
 2. Carga elétrica (ex: cromatografia de troca iônica, eletroforese) 3. Solubilidade ou hidrofobicidade (ex: cromatografia em papel, fase reversa) Métodos baseados em afinidade biológica, que exploram a interação entre duas. moléculas: 4. Cromatografia de afinidade (pressupõe que uma das moléculas do par que. interage é um reagente de fácil obtenção, disponível comercialmente)")
4
. . . . . . 1 Proteína apenas MATERIAL BIOLÓGICO DE PARTIDA: (100g)
ORGANELAS Lisossomos Mitocôndria Golgi Núcleo SOBRENADANTE F 1 2 3 4 .1 .2 .3 .3.1 .3.2 2. 3.N Precipitação com Sal/Solvente Cromatografia Troca Iônica (pH ou resina diferente) .3.N.X 1 Proteína apenas (0.001g) a 0.5% total Gel Filtração MATERIAL BIOLÓGICO DE PARTIDA: (100g) O fluxograma ao lado representa a “marcha” de purificação de uma proteína, mostrando as etapas sucessivas, cada uma consistindo de método, que levam ao isolamento de uma proteína presente numa mistura complexa. Observe a quantidade de proteínas (100g) presente no material inicial e quanto da proteína purificada se obtém no final (0,001g). Esses números são típicos para a purificação da maioria das proteínas, em especial enzimas. Em cada etapa, a proteína de interesse é separada das demais com base em uma propriedade diferente. Consequente-mente, as proteínas ainda misturadas com aquela que está sendo isolada são cada vez mais semelhantes em suas características físico-químicas, exigindo métodos cada mais sensíveis, capazes de explorar pequenas diferenças, para se chegar à proteína pura.
 .3.N.X. 1 Proteína apenas. (0.001g) a 0.5% total. Gel Filtração. MATERIAL BIOLÓGICO DE PARTIDA: (100g) O fluxograma ao lado representa a marcha de purificação de uma proteína, mostrando as etapas sucessivas, cada uma consistindo de método, que levam ao isolamento de uma proteína presente numa mistura complexa. Observe a quantidade de proteínas (100g) presente no material inicial e quanto da proteína purificada se obtém no final (0,001g). Esses números são típicos para a purificação da maioria das proteínas, em especial enzimas. Em cada etapa, a proteína de interesse é separada das demais com base em uma propriedade diferente. Consequente-mente, as proteínas ainda misturadas com aquela que está sendo isolada são cada vez mais semelhantes em suas características físico-químicas, exigindo métodos cada mais sensíveis, capazes de explorar pequenas diferenças, para se chegar à proteína pura.")
5
. . . . . . 1 Proteína apenas MATERIAL BIOLÓGICO DE PARTIDA: (100g)
ORGANELAS Lisossomos Mitocôndria Golgi Núcleo SOBRENADANTE F 1 2 3 4 .1 .2 .3 .3.1 .3.2 2. 3.N Precipitação com Sal/Solvente Cromatografia Troca Iônica (pH ou resina diferente) .3.N.X 1 Proteína apenas (0.001g) a 0.5% total Gel Filtração MATERIAL BIOLÓGICO DE PARTIDA: (100g) Além dos métodos de purificação, análises complementares devem ser feitas ao longo da purificação, para verificar se o processo de separação está sendo eficiente. A medida da atividade biológica da proteína de interesse e do conteúdo de proteínas de cada fração resultante do processo de separação devem ser feitas a cada passo. Assim, apenas a fração que contém a proteína de interesse, marcada com um círculo vermelho no fluxograma, é submetida à próxima etapa de purificação. Medida da atividade biológica - particular para cada proteína - deve ser quantitativa, para estimar quanto da proteína de interesse há em cada fração. Medidas do conteúdo proteico (diversos) - absorbância de luz UV de 280 nm - métodos colorimétricos (ex: Lowry, Bradford, BCA, etc)
 .3.N.X. 1 Proteína apenas. (0.001g) a 0.5% total. Gel Filtração. MATERIAL BIOLÓGICO DE PARTIDA: (100g) Além dos métodos de purificação, análises complementares devem ser feitas ao longo da purificação, para verificar se o processo de separação está sendo eficiente. A medida da atividade biológica da proteína de interesse e do conteúdo de proteínas de cada fração resultante do processo de separação devem ser feitas a cada passo. Assim, apenas a fração que contém a proteína de interesse, marcada com um círculo vermelho no fluxograma, é submetida à próxima etapa de purificação. Medida da atividade biológica. - particular para cada proteína. - deve ser quantitativa, para estimar quanto da proteína de interesse há em cada fração. Medidas do conteúdo proteico (diversos) - absorbância de luz UV de 280 nm. - métodos colorimétricos. (ex: Lowry, Bradford, BCA, etc)")
6
Métodos para medida do conteúdo de proteína:
Comprimento de onda Absortividade molar, e A absorbância de uma solução de proteínas a 280 nm é diretamente proporcional ao seu conteúdo proteico, desde que essas contenham esses aminoácidos aromáticos na sua composição, especialmente triptofano. A vantagem desse método é que ele não é destrutivo para a proteína. Em geral, considera-se que uma leitura de 1,0 A280 equivale a uma concentração de 1 mg/mL. Ácidos nucleicos também absorvem luz UV – máxomo 260 nm. Proteína: razão A280/A260 > 1

7
Métodos para medida do conteúdo de proteína:
Reagente de Biureto: sulfato de Cu2+ em tartarato Cu2+ reage com as ligações peptídicas e produz um complexo púrpura com absorção máxima em 540 nm 1. Cu2+ é chelado pela proteína [Cu2+-proteina] 2. Reação redox Cu2+ + (ligações peptídicas) —> [Cu+ -proteína] Reagente Bradford = Coomassie Brilliant Blue G-250 Método baseado em uma mudança espectral do reagente, em que dá absorção máxinma a 595 nm (cor azul), quando interage com proteínas. Interação com proteínas se dá através de forças de Van der Waals e ligações iônicas, especialmente com arginina, mas também com resíduos de histidina, lisina, tirosina, triptofano e fenlialanina. Método de Lowry Ìons Cu2+ reduzidos a Cu+ em presença de proteínas e cadeias laterais de aminoácidos aromáticos, Tyr e Trp, e de Cys, reduzem o reagente de Folin-Ciocalteu (ácido fosfo- mobidênio-tungstênio), dando um composto de cor azul. BCA = ácido 2,2'-Bicinchonínico ou 4,4'-Dicarboxy-2,2'-biquinoline Ìons Cu2+ reduzidos a Cu+ em presença de proteínas reagem fortemente com o BCA formando um composto azul,
 —> [Cu+ -proteína] Reagente Bradford = Coomassie Brilliant Blue G-250. Método baseado em uma mudança espectral do reagente, em que dá absorção máxinma a 595 nm (cor azul), quando interage com proteínas. Interação com proteínas se dá através de forças de Van der Waals e ligações iônicas, especialmente com arginina, mas também com resíduos de histidina, lisina, tirosina, triptofano e fenlialanina. Método de Lowry. Ìons Cu2+ reduzidos a Cu+ em presença de proteínas e cadeias laterais de aminoácidos aromáticos, Tyr e Trp, e de Cys, reduzem o reagente de Folin-Ciocalteu (ácido fosfo- mobidênio-tungstênio), dando um composto de cor azul. BCA = ácido 2,2 -Bicinchonínico ou 4,4 -Dicarboxy-2,2 -biquinoline. Ìons Cu2+ reduzidos a Cu+ em presença de proteínas reagem fortemente com o BCA formando um composto azul,")
8
(para romper tecidos e células)
Para iniciar a purificação, inicialmente é necessário extrair a proteína de interesse para um meio líquido, exceto se ela já estiver naturalmente presente em um meio líquido (sangue, suor, água do mar, meio de cultura, seiva de planta, etc). tecidos células Homogeneização (para romper tecidos e células) por pressão (prensa francesa) liquefação (Potter) Homogenado ou Extrato bruto Material de partida Material na fonte por ultra-som com detergente Vários métodos são possíveis para transformar células, órgãos, tecidos em um homogenado, ou extrato bruto, como mostra a figura.
. tecidos. células. Homogeneização. (para romper tecidos e células) por pressão. (prensa francesa) liquefação. (Potter) Homogenado. ou. Extrato bruto. Material de partida. Material na. fonte. por ultra-som. com. detergente. Vários métodos são possíveis para transformar células, órgãos, tecidos em um homogenado, ou extrato bruto, como mostra a figura.")
9
Detergentes podem ser iônicos e não iônicos
Proteínas integrais da membrana detergente micelas Complexos não micelares Dependendo da concentração Sendo a proteína de interesse uma proteína de membrana, é necessário solubilizá-la. Para esse fim são utilizados detergentes, que dissolvem a membrana plasmática e formando complexos solúveis com as proteínas. Detergentes podem ser iônicos e não iônicos Detergentes não iônicos Triton X-100 (polyoxyethylene (9,5) p-t-octylphenol) SDS Dodecil sulfato de sódio Cetab Cetyltrimethylammonium bromide Deoxicolato de sódio Detergentes iônicos Octilglicosídeo (octyl-b-D-glucopyranoside)
 p-t-octylphenol) SDS. Dodecil sulfato de sódio. Cetab. Cetyltrimethylammonium bromide. Deoxicolato de sódio. Detergentes iônicos. Octilglicosídeo. (octyl-b-D-glucopyranoside)")
10
A centrífuga centrifugação diferencial
Câmara blindada Amostra sedimentando refrigeração vácuo rotor ângulo fixo A centrifugação frequentemente é uma das primeiras etapas de purificação aplicado a um extrato bruto. Através do movimento de rotação do rotor da centrífuga, uma força centrífuga é aplicada à amostra, separando seus compo-nentes através de suas massas e/ou densidade, conforme a técnica. Através de sucessivas etapas de centrifução com velocidades (rotações por minuto, rpm) crescentes, pode-se obter diferentes frações de um homogenado de células ou tecidos. centrifugação diferencial homogenado células intactas pedaços de membrana núcleos mitocôndrias lisossomos ribossomos fração citoplasmática sangue centrifugado: separação de plasma e células
 crescentes, pode-se obter diferentes frações de um homogenado de células ou tecidos. centrifugação diferencial. homogenado. células intactas. pedaços de membrana. núcleos. mitocôndrias. lisossomos. ribossomos. fração. citoplasmática. sangue centrifugado: separação de plasma e células.")
11
Força centrífuga Centrífuga Clínica Ultracentrífuga R$ 3.000
Existem centrífugas para diferentes tipos de aplicações, dependendo da força centrífuga que são capazes de gerar. Relação entre o raio do rotor, velocidade angular (rpm) e a força centrífuga (g) Centrífuga Clínica Ultracentrífuga (necessitam vácuo para evitar atrito do ar, além de refrigeração) 1,200g por 15 minutos separa plasma de células sanguíneas 200,000 g por 24 horas para separar organelas menores ou complexos proteicos R$ 3.000 US$ 70,000 Preço aproximado
 e a força centrífuga (g) Centrífuga. Clínica. Ultracentrífuga. (necessitam vácuo para evitar atrito do ar, além de refrigeração) 1,200g por 15 minutos separa plasma de células sanguíneas. 200,000 g por 24 horas para separar organelas menores ou complexos proteicos. R$ US$ 70,000. Preço aproximado.")
12
Centrifugação em gradiente de densidade
A centrifugação em um meio com gradiente de densidade melhora a eficiência da separação. As partículas se deslocarão através do gradiente até encontrarem uma região com densidade equivalente a sua, quando param de se mover, formando “bandas”. Gradientes podem ser utilizados para separar diferentes tipos de células, organelas, ácidos nucléicos, complexos proteicos, etc. Centrifugação em gradiente de densidade Solucões de sacarose com densidades diferentss são colocadas no tubo, uma sobre a outra Amostra é colocada no topo do gradiente 5% sacarose 20% sacarose centrifugação Baixa densidade Média densidade Alta densidade Gradiente separação de: Ficoll-Hypaque leucócitos Sacarose mitocôndrias Cloreto de césio ácidos nucléicos

13
A precipitação de proteínas pode ser induzida por:
As mais potentes ultracentrífugas atuais ainda não são capazes de sedimentar proteínas. Processos que diminuem a solubilidade e provocam a precipitação fracionada de proteínas são utilizados como etapas preliminares de purificação. Uma das vantagens desses métodos é o baixo custo e capacidade de processar grandes volumes/massas. A precipitação de proteínas pode ser induzida por: - adição de sais (Precipitação salina) - adição de solventes - variação de pH (Precipitação isoelétrica) O gráfico ao lado ilustra o efeito de diferentes sais sobre a solubilidade da hemoglobina. Em baixa concentração salina, a solubilidade das proteínas aumenta, pois os íons do sal ajudam a reforçar a camada de solvatação. Em alta concentração salina, a solubilidade das proteínas diminue pois os íons do sal competem pelas moléculas de água disponíveis para formar a sua própria camada de solvatação. Sais com ânions divalentes são mais eficientes do que os monovalentes na precipitação de proteínas. Solubilidade da hemoglobina (S/S’) KCl NaCl MgSO4 (NH4)2SO4 K2SO4 Concentração do Sal,, Molar Salting-in X Salting-out
 - adição de solventes. - variação de pH (Precipitação isoelétrica) O gráfico ao lado ilustra o efeito de diferentes sais sobre a solubilidade da hemoglobina. Em baixa concentração salina, a solubilidade das proteínas aumenta, pois os íons do sal ajudam a reforçar a camada de solvatação. Em alta concentração salina, a solubilidade das proteínas diminue pois os íons do sal competem pelas moléculas de água disponíveis para formar a sua própria camada de solvatação. Sais com ânions divalentes são mais eficientes do que os monovalentes na precipitação de proteínas. Solubilidade da hemoglobina (S/S’) KCl. NaCl. MgSO4. (NH4)2SO4. K2SO4. Concentração do Sal,, Molar. Salting-in X Salting-out.")
14
- precipitam primeiro: proteínas maiores proteínas mais hidrofóbicas
Sais se dissociam em solucão aquosa e competem com as proteínas pela água de solvatação. Solubilidade, log Fibrinogênio Pseudoglobulina Mioglobina Albumina Hemoglobina Concentração do Sal (NH4)2SO4,, Molar Considere uma solução com fibrinogênio, albumina, hemoglobina, pseudoglobulina e mioglobina e observe o gráfico. Usando o sal sulfato de amônio (NH4)2SO4, pode-se purificar completamente o fibrinogênio a partir de uma mistura das 5 proteínas, pois este precipita totalmente em uma saturação de 2,8 M do sal. Neste ponto, centrifuga-se a solução e o fibrinogênio é coletado como um precipitado. Numa próxima etapa, adicionando-se mais sal à solução até uma saturação de 7 M, pode-se obter um novo precipitado contendo albumina, hemoglobina e pseudoglobulina. E teremos também purificada a mioglobina, ainda em solúvel na presença de 7M do sal, e que ficou sozinha no sobrenadante. Proteínas apresentam diferente sensibilidade para a precipitação salina. - precipitam primeiro: proteínas maiores proteínas mais hidrofóbicas possuem camadas de solvatação maiores ou menos organizadas, mais fáceis de pertubar.
2SO4,, Molar. Considere uma solução com fibrinogênio, albumina, hemoglobina, pseudoglobulina e mioglobina e observe o gráfico. Usando o sal sulfato de amônio (NH4)2SO4, pode-se purificar completamente o fibrinogênio a partir de uma mistura das 5 proteínas, pois este precipita totalmente em uma saturação de 2,8 M do sal. Neste ponto, centrifuga-se a solução e o fibrinogênio é coletado como um precipitado. Numa próxima etapa, adicionando-se mais sal à solução até uma saturação de 7 M, pode-se obter um novo precipitado contendo albumina, hemoglobina e pseudoglobulina. E teremos também purificada a mioglobina, ainda em solúvel na presença de 7M do sal, e que ficou sozinha no sobrenadante. Proteínas apresentam diferente sensibilidade para a precipitação salina. - precipitam primeiro: proteínas maiores. proteínas mais hidrofóbicas. possuem camadas de solvatação maiores. ou menos organizadas, mais fáceis de pertubar.")
15
Os mais utilizados são etanol e acetona.
Proteínas também podem ser precipitadas com adição de solventes ao meio ou quando colocadas em meio com pH próximo ao seu ponto isoelétrico. Proteína P.I. Pepsina <1,0 Ovalbumina galinha 4,6 Albumina sérica humana 4,9 Tropomiosina 5,1 Insulina bovina 5,4 Fibrinogênio humano 5,8 Gama-globulina 6,6 Colágeno 6,6 Mioglobina equina 7,0 Hemoglobina humana 7,1 Ribonuclease A bovina 7,8 Citocromo C equino 10,6 Histona bovina 10,8 Lisozima, galinha 11,0 Salmina, salmão 12,1 Proteínas colocadas em meio com pH igual ao seu PI tendem a precipitar, pois tendo carga neutra, apresentam a camada de solvatação menos organizada. Água Dimetilformamida Metanol Etanol Acetona Clorofórmio Benzeno 78.5 1.85 48.9 3.96 32.6 1.66 20.7 2.72 2.3 0.00 Solvente Constante Dielétrica Momento Dipolar Solventes miscíveis com a água diminuem a constante dielétrica do meio e desorganizam a camada de solvatação das proteínas. Os mais utilizados são etanol e acetona. Ponto Isoelétrico de algumas Proteínas

16
Para obter as proteínas precipitadas em solução novamente, é necessário reverter as condições que levaram à precipitação. Aos precipitados obtidos com sal ou solvente, adiciona-se água. Ao precipitado obtido com variação de pH, retornar ao pH original. Membrana de celofane solvente solução a ser dialisada início final Dt Para remover o excesso de sal ou do solvente no precipitado, utiliza-se a diálise. amostra é colocada dentro de um saco feito com uma membrana de celofane com poros tratados, que permite a passagem de moléculas até 10,000 d. - o saco contendo a amostra é imerso em um recipiente contendo o solvente que se deseja - excesso de sal ou de solvente se difunde para o solvente - após várias trocas de solvente, todo o excesso de sal/solvente terá sido retirado.

17
Até o momento, exploramos as etapas iniciais de purificação de proteínas, como a centrifugação diferencial e precipitação fracionada. Esses métodos, apesar de terem baixo poder de resolução (exploram diferenças grosseiras entre as proteínas), permitem processar grandes volumes ou massas, típicos das etapas iniciais de isolamento. Quando já houve redução significa-tiva dos montantes de proteínas a serem processados, iniciam-se as cromatografias, que irão explorar as diferenças mais sutis entre as moléculas. Não esquecer que todas as frações obtidas devem ter o conteúdo de proteínas e de atividade biológica medidos, para se decidir qual/quais deverão passar para o passo seguinte da purificação. ORGANELAS Lisossomos Mitocôndria Golgi Núcleo SOBRENADANTE F 1 2 3 4 .1 .2 .3 .3.1 .3.2 2. 3.N Precipitação com Sal/Solvente Cromatografia Troca Iônica (pH ou resina diferente) .3.N.X 1 Proteína apenas (0.001g) a 0.5% total Gel Filtração MATERIAL BIOLÓGICO DE PARTIDA: (100g)
, permitem processar grandes volumes ou massas, típicos das etapas iniciais de isolamento. Quando já houve redução significa-tiva dos montantes de proteínas a serem processados, iniciam-se as cromatografias, que irão explorar as diferenças mais sutis entre as moléculas. Não esquecer que todas as frações obtidas devem ter o conteúdo de proteínas e de atividade biológica medidos, para se decidir qual/quais deverão passar para o passo seguinte da purificação. ORGANELAS. Lisossomos. Mitocôndria. Golgi. Núcleo. SOBRENADANTE. F N. Precipitação com. Sal/Solvente. Cromatografia Troca Iônica. (pH ou resina diferente) .3.N.X. 1 Proteína apenas. (0.001g) a 0.5% total. Gel Filtração. MATERIAL BIOLÓGICO DE PARTIDA: (100g)")
18
Que características devem ter as resina cromatográficas para possibilitar separações de moléculas baseadas em diferentes propriedades ? Cromatografia de permeação em gel ou gel-filtração: separação de moléculas pela massa molecular géis são porosos, funcionando como peneiras ou filtros Cromatografia de troca iônica: separação de moléculas pela carga elétrica géis apresentam grupos carregados positiva- ou negativamente Cromatografia de partição (fase reversa ou hidrofóbica): separação de moléculas pela solubilidade relativa em meio aquoso géis possuem carácter hidrofóbico Cromatografia de afinidade: separação de moléculas pela capacidade de interagir com um ligante géis possuem ligante específico ligado covalente à resina
: separação de moléculas pela solubilidade relativa em meio aquoso. géis possuem carácter hidrofóbico. Cromatografia de afinidade: separação de moléculas pela capacidade de interagir com um ligante géis possuem ligante específico ligado covalente à resina.")
19
Como se avalia se o processo de purificação de uma proteína foi eficiente ?
Três parâmetros permitem avaliar a eficiência do processo de purificação de uma proteína: Atividade específica (AE): é a razão entre a quantidade da proteína de interesse, medida através de sua atividade biológica, e a quantidade total de proteínas presentes em cada etapa de purificação. Esse índice aumenta ao longo da purificação. Índice de purificação: indica quantas vezes em relação ao material de partida a proteína de interesse foi “concentrada”. Calcula-se como a razão entre as atividades específicas inicial (material de partida) e final (proteína pura). Rendimento: indica quanto (em %) da proteína de interesse ativa presente no material de partida foi recuperado ao final da purificação. Um certo grau de perda é inerente do processo de purificação (em cada etapa só devem ser proces sadas as frações mais ricas em atividade biológica, desprezando-se aquelas que apresentam pouca atividade). Também podem ocorrer perdas por desnaturação das proteínas devido às diferentes condições (pH, sais, etc) a que são submetidas nas diferentes etapas de purificação. Espera-se recuperar o máximo possível da proteína de interesse.
: é a razão entre a quantidade da proteína de interesse, medida. através de sua atividade biológica, e a quantidade total de proteínas presentes. em cada etapa de purificação. Esse índice aumenta ao longo da purificação. Índice de purificação: indica quantas vezes em relação ao material de partida a proteína. de interesse foi concentrada . Calcula-se como a razão entre as atividades. específicas inicial (material de partida) e final (proteína pura). Rendimento: indica quanto (em %) da proteína de interesse ativa presente no material de. partida foi recuperado ao final da purificação. Um certo grau de perda é inerente do processo de purificação (em cada etapa só devem ser proces- sadas as frações mais ricas em atividade biológica, desprezando-se aquelas. que apresentam pouca atividade). Também podem ocorrer perdas por desnaturação das proteínas devido às diferentes condições (pH, sais, etc) a que são submetidas nas diferentes. etapas de purificação. Espera-se recuperar o máximo possível da proteína de interesse.")
20
Purificação da Glicoquinase hepática de Rato
Etapa Atividade Específica b ( U ( U.mg-1) . - a Rendimento c ( % ) 0.17 2.0 85 12 23 45 140 Marcha A: somente cromatografias convencionais 1. Sobrenadante de pâncreas 2. (NH4) 2 SO4, precipitado 25 -55% 3. troca iônica, coluna DEAE -Sephadex , pH 7.0, eluição KCl 4. troca iônica, coluna CM cellulose , pH 5.5, NaCl 6. interação hidrofóbica, coluna Butyl~Sepharose 7. gel filtração, coluna Sephacryl S -300 130 Marcha B: com cromatografia de afinidade 0.09 2. troca iônica, coluna DEAE Sephadex 3. Afinidade cromatográfica ( benzamidina~agarose ) Índice Purificação a U nidade de enzima (1 unidade de atividade enzimática é definida como a quantidade de enzima que cataliza a transformação de 1 micromol de substrato em produto, em condições pre-estabelecidas b Atividade específica : medida da atividade biológica (em U) expressa por miligramas de proteína c Rendimento : percentual da atividade biológica inicial (100%) recuperada em cada etapa de purificação d Índice de Purificação : razão entre a atividade específica inicial e aquela determinada em cada etapa.
 . - a. Rendimento. c. ( % ) Marcha A: somente cromatografias convencionais. 1. Sobrenadante. de pâncreas. 2. (NH4) 2. SO4, precipitado % 3. troca iônica, coluna DEAE. -Sephadex. , pH 7.0, eluição. KCl. 4. troca iônica, coluna. CM. cellulose. , pH 5.5, NaCl. 6. interação hidrofóbica, coluna. Butyl~Sepharose. 7. gel. filtração, coluna. Sephacryl. S Marcha B: com cromatografia de afinidade troca iônica, coluna DEAE. Sephadex Afinidade cromatográfica ( benzamidina~agarose. ) Índice. Purificação. a. U. nidade de enzima (1 unidade de atividade enzimática é definida como a quantidade de enzima que cataliza. a transformação de 1 micromol de substrato em produto, em condições pre-estabelecidas. b. Atividade específica. : medida da atividade biológica (em U) expressa por miligramas de proteína. c. Rendimento. : percentual da atividade biológica inicial (100%) recuperada em cada etapa de purificação. d. Índice de Purificação. : razão entre a atividade específica inicial e aquela determinada em cada etapa.")
21
Funcionamento Básico de uma Coluna Cromatográfica
Uma coluna é um tubo cilindríco aberto nas duas extremidades e preenchido com a resina ou matriz ou gel cromatográfico. A coluna é constantemente alimentada com líquido (tampão), banhando a resina e forçando o contacto desta e as moléculas que estão sendo analisadas. Abaixo está representado o esquema geral de uma cromatografia: Amostra com diferentes componentes Resina embebida em tampão Tempo zero Concentração Tempo ou volume Coletor de frações Tempo 1 Mais tampão é colocado na coluna, forçando os componentes da amostra a interagirem com a resina Componentes da amostra se separam e saem da coluna com diferentes volumes de tampão Tempo 2 Tempo n Líquido que sae da coluna é recolhido em tubos de um coletor de frações Os componentes da mistura são separados por interação diferenciada com a resina, com base em propriedades moleculares como: massa molecular carga elétrica solubilidade afinidade Um cromatograma, como o gráfico ao lado, é a maneira usual de se representar o resultado de uma cromatografia.
, banhando a resina e forçando o contacto desta e as moléculas que estão sendo analisadas. Abaixo está representado o esquema geral de uma cromatografia: Amostra com diferentes componentes. Resina embebida em tampão. Tempo zero. Concentração. Tempo ou volume. Coletor de frações. Tempo 1. Mais tampão é colocado na coluna, forçando os componentes da amostra a interagirem com a resina. Componentes da amostra se separam e saem da coluna com diferentes volumes de tampão. Tempo 2. Tempo n. Líquido que sae da coluna é recolhido em tubos de um coletor de frações. Os componentes da mistura são separados por interação diferenciada com a resina, com base em propriedades moleculares como: massa molecular. carga elétrica. solubilidade. afinidade. Um cromatograma, como o gráfico ao lado, é a maneira usual de se representar o resultado de uma cromatografia.")
22
Cromatografia de gel filtração ou peneira molecular
Moléculas com massas diferentes grão da resina poroso proteína grande proteína pequena Tampão “empurra” moléculas através da resina tubos Fluxo do tampão grãos (beads) da resina com poros Na gel-filtração, as proteínas que penetram nos poros da resina precisam diferentes volumes de tampão para saírem da coluna, conforme suas massas moleculares, percorrendo os canais internos dos grãos. Quanto maior o número de grãos que cada molécula entrar durante o percurso através da coluna, maior o volume necessário para sua saída. Proteínas maiores que o diâmetro dos poros não são separadas e saem da coluna com pouco tampão, correspondente apenas ao volume da coluna externo aos grãos, também chamado de volume morto (Vo). Proteínas menores que o diâmetro dos poros não são separadas e saem da coluna com um volume de tampão correspondente ao volume interno (Vi, volume total menos o volume do próprio gel).
 da resina com poros. Na gel-filtração, as proteínas que penetram nos poros da resina precisam diferentes volumes de tampão para saírem da coluna, conforme suas massas moleculares, percorrendo os canais internos dos grãos. Quanto maior o número de grãos que cada molécula entrar durante o percurso através da coluna, maior o volume necessário para sua saída. Proteínas maiores que o diâmetro dos poros não são separadas e saem da coluna com pouco tampão, correspondente apenas ao volume da coluna externo aos grãos, também chamado de volume morto (Vo). Proteínas menores que o diâmetro dos poros não são separadas e saem da coluna com um volume de tampão correspondente ao volume interno (Vi, volume total menos o volume do próprio gel).")
23
traçado da medida de atividade biológica nas frações
A cromatografia de gel-filtração possibilita estimar a massa molecular de uma proteína em seu estado nativo Além de purificar, por ser realizada em condições de pH, força iônica e temperatura que preservam a atividade biológica da proteína, a gel-filtração fornece a Mr do seu estado nativo. Para isso, é necessário “calibrar” a coluna com proteínas de massa molecular conhecida, construindo-se uma curva de calibração. O volume de saída (eluição) de uma proteína numa coluna de gel-filtração é proporcional ao logaritmo de sua massa molecular. Curva de calibração Observar a escala log traçado da medida de atividade biológica nas frações Moléculas maiores Moléculas menores Massa molecular (kD) Absorbância a 280 nm Ler a massa correspondente Medida da ativ. biológica mL mL volume de eluição Medir o volume de eluição da fração mais ativa. Transportar para a curva de calibraçao. volume Kav
 de uma proteína numa coluna de gel-filtração é proporcional ao logaritmo de sua massa molecular. Curva de calibração. Observar a escala log. traçado da medida de atividade biológica nas frações. Moléculas maiores. Moléculas menores. Massa molecular (kD) Absorbância a 280 nm. Ler a massa correspondente. Medida da ativ. biológica mL mL. volume de eluição. Medir o volume de eluição da fração mais ativa. Transportar para a curva de calibraçao. volume. Kav.")
24
Resinas para Gel-Filtração
Existem diferentes tipos de resinas para gel-filtração, conforme o tipo de moléculas ou partículas a serem separadas indica quais os tamanhos das partículas que podem entrar nos poros da resina e serem fracionados. Acima ou abaixo da faixa, não há separação. Resinas para Gel-Filtração Sephadex G - 10 Dextrana 0.05 0.70 25 1 5 50 30 100 4 150 200 600 Bio Gel P 2 Policrilamida 0.1 1.8 6 1.5 20 2.4 40 300 60 400 Sepharose 6B Agarose 4.000 Sepharose 4B 20.000 Sepharose 2B 70 40.000 NOME TIPO FAIXA DE RESOLUÇÃO (kD) *Sephadex e Sepharose: Amersham Pharmacia Biotech; Bio Gel: Bio Rad Laboratories Moléculas pequenas Proteínas Moléculas pequenas Proteínas Células, partículas sub-celulares
 *Sephadex e Sepharose: Amersham Pharmacia Biotech; Bio. Gel: Bio. Rad Laboratories. Moléculas pequenas. Proteínas. Moléculas pequenas. Proteínas. Células, partículas sub-celulares.")
25
Esta é uma outra forma de representar a calibração de uma coluna de gel-filtração.
O gel nessa coluna é o Sephadex G-200, cuja faixa de resolução de proteínas é de a d. Observe como proteínas nos extremos da faixa de resolução tendem a “sair” da parte linear da curva. Para comparar calibrações com a mesma resina cromatográfica em colunas de dimensões diferentes utiliza-se o Kav, que é proporcional ao log de Mr. Kav = Ve-Vo Vt-Vo Ve – volume de eluição de uma certa proteína Vt – volume total da coluna Vo – volume morto da coluna, em que saem moléculas com massa acima da resolução da resina onde:

26
Cromatografia de troca iônica
A resina para cromatografia de troca iônica apresenta carga elétrica, positiva ou negativa, em uma ampla faixa de pH. Trocadora de ânions Trocadora de cátions DEAE CM Existem dois tipos básicos: resinas trocadoras de ânions (possuem carga positiva), como o dietilaminoetil (DEAE)-celulose e resinas trocadoras de cátions (possuem carga negativa), como o carboxi-metil (CM)-celulose
, como o dietilaminoetil (DEAE)-celulose e resinas trocadoras de cátions (possuem carga negativa), como o carboxi-metil (CM)-celulose.")
27
- Cromatografia de troca iônica + -
Ponto Isoelétrico de algumas Proteínas Proteína P.I. Pepsina <1,0 Ovalbumina galinha 4,6 Albumina sérica humana 4,9 Tropomiosina 5,1 Insulina bovina 5,4 Fibrinogênio humano 5,8 Gama-globulina 6,6 Colágeno 6,6 Mioglobina equina 7,0 Hemoglobina humana 7,1 Ribonuclease A bovina 7,8 Citocromo C equino 10,6 Histona bovina 10,8 Lisozima, galinha 11,0 Salmina, salmão 12,1 DEAE-celulose - pH < 10 CM-celulose - pH > 4 PI ácido + básico - neutro

28
Como funciona a Cromatografia de Troca Iônica
Adsorção No exemplo ao lado, como funciona uma resina catiônica ou trocadora de ânions, como o DEAE-celulose): + A cromatografia de troca iônica compreende duas etapas: adsorção das proteínas com carga contrária à resina, e saída da coluna das proteínas com a mesma carga; eluição das proteínas adsorvidas. Moléculas com a mesma carga, ou sem carga, não interagem com a resina, sendo as primeiras a sair da coluna Eluição Para a eluição, as condições de adsorção da coluna (pH ou força iônica) são alteradas para neutralizar a interação entre as proteínas e a resina. Mais frequentemente utiliza-se um aumento da concentração do sal no tampão, pois alterações de pH podem desnaturar proteínas, levando-as a precipitar dentro da coluna. + Na+Cl- + Adição de sal ao tampão resulta em competição entre os íons em solução e as moléculas adsorvidas na resina.
: + A cromatografia de troca iônica compreende duas etapas: adsorção das proteínas com carga contrária à resina, e saída da coluna das proteínas com a mesma carga; eluição das proteínas adsorvidas. Moléculas com a mesma carga, ou sem carga, não interagem com a resina, sendo as primeiras a sair da coluna. Eluição. Para a eluição, as condições de adsorção da coluna (pH ou força iônica) são alteradas para neutralizar a interação entre as proteínas e a resina. Mais frequentemente utiliza-se um aumento da concentração do sal no tampão, pois alterações de pH podem desnaturar proteínas, levando-as a precipitar dentro da coluna. + Na+Cl- + Adição de sal ao tampão resulta em competição entre os íons em solução e as moléculas adsorvidas na resina.")
29
DEAE CM Duas Modalidades de Cromatografia de Troca Iônica Eluição NaCl
Gradientes de sal podem fracionar as proteínas adsorvidas na resina de acordo com a intensidade de suas cargas, que é dada pela diferença entre seus PIs e o pH do tampão de eluição. As proteínas não retidas (com a mesma carga da resina) não são separadas, sendo simplesmente “arrastadas” pelo tampão (ou seja, não são repelidas pela resina). _ = + (+) 0. 10 M 0. 15 M 0. 20 M Eluição NaCl Não retidas DEAE + 0. 10 M 0. 15 M 0. 20 M Eluição NaCl (-) _ = Não retidas CM Carboximetil~celulose (trocadora de cátions) Dietilaminoetil~celulose (trocadora de ânions)
 não são separadas, sendo simplesmente arrastadas pelo tampão (ou seja, não são repelidas pela resina). _. = + (+) M M M. Eluição NaCl. Não retidas. DEAE M M M. Eluição NaCl. (-) _. = Não retidas. CM. Carboximetil~celulose. (trocadora de cátions) Dietilaminoetil~celulose. (trocadora de ânions)")
30
Tipos de Resina de troca iônica
NOME TIPO GRUPO IONIZÁVEL OBSERVAÇÕES Dowex 1 Fortemente básica Ø - CH 2 N+(CH 3 ) Troca aniônica resina de polistireno Dowex 50 SO -H Troca Catiônica DEAE celulose Básica Dietilaminoetil Fracionamento de proteínas N+(C H 5 ácidas e neutras CM Ácida Carboximetil COOH básicas e neutras Q-Sepharose Gel de dextrano Combinação de gel filtração básico e troca iônica de proteínas SP -Sepharose ácido * Dowex: Dow Chemical Co.; Sepharose e Source: GE LifeSciences Bio Gel: Bio Rad Laboratories Tipos de Resina de troca Iônica −Ο−CH2CHOHCH2OCH2CHOHCH2N+(CH3)3 —CH2-SO3- Existem vários tipos de resinas de troca iônica disponíveis no mercado.
 Troca aniônica. resina de polistireno. Dowex 50. SO. -H. Troca Catiônica. DEAE. celulose. Básica. Dietilaminoetil. Fracionamento de proteínas. N+(C. H. 5. ácidas e neutras. CM. Ácida. Carboximetil. COOH. básicas e neutras. Q-Sepharose. Gel de dextrano. Combinação de gel filtração. básico. e troca iônica de proteínas. SP. -Sepharose. ácido. * Dowex: Dow Chemical Co.; Sepharose e Source: GE LifeSciences. Bio. Gel: Bio Rad Laboratories. Tipos de Resina de troca Iônica. −Ο−CH2CHOHCH2OCH2CHOHCH2N+(CH3)3. —CH2-SO3- Existem vários tipos de resinas de troca iônica disponíveis no mercado.")
31
Cromatografia de afinidade:
Ex: cromatografia de imunoafinidade Um dos métodos mais eficientes para a purificação de proteínas, possibilitando um alto rendimento com número reduzido de etapas. A separação de moléculas tem como base a interação específica do analito (molécula-alvo) com um ligante imobilizado na matriz. Forças envolvidas nessa interação podem ser não covalentes (eletrostáticas, hidrofóbica, pontes de H) ou covalentes (p.e., ponte dissulfeto). Desprezar proteínas não retidas Lavagem Eluição pH 2,0 ou sal Antígeno A puro Anti-A interage apenas com a proteína A - Mistura de proteínas Coluna empacotada com ess gel Partículas de gel recobertas de anticorpos anti-A Adsorção Complexo Ag-AC é desfeito Eluição: condições que interferem na ligação da proteína ao ligante, como mudanças no pH e/ou força iônica, ou por competição com o ligante livre
 com um ligante imobilizado na matriz. Forças envolvidas nessa interação podem ser não covalentes (eletrostáticas, hidrofóbica, pontes de H) ou covalentes (p.e., ponte dissulfeto). Desprezar proteínas não retidas. Lavagem. Eluição. pH 2,0 ou sal. Antígeno A puro. Anti-A interage apenas com a proteína A. - Mistura de proteínas. Coluna empacotada com ess gel. Partículas de gel recobertas de anticorpos anti-A. Adsorção. Complexo Ag-AC é desfeito. Eluição: condições que interferem na ligação da proteína ao. ligante, como mudanças no pH e/ou força iônica, ou. por competição com o ligante livre.")
32
Tipo de ligantes em cromatografia de afinidade
1. Mono-específicos - ligação específica da molécula-alvo - análogos de substratos ou inibidores de enzimas - agonistas ou antagonistas de receptores - haptenos ou determinantes antigênicos de anticorpos - ligantes com “tag” ou “marcação” - glutationa-S-transferase - poli-Histidina 2. Grupo-específico: ligantes para separação de grupos: 8-AEA-cAMP 8-(2-aminoethyl)aminoadenosine-3',5'-cyclic monophosphate (ligante para proteínas com afinidade por cAMP ou cGMP) Ligante grupo-específico Especificidade Proteína A Região Fc de IgG Proteína G Concanavalina A Grupos glicosil- ou manosil- Cibacron Blue Várias enzimas, albumina lisina Plasminogênio, RNA ribossomal arginina proteinases tipo tripsina benzamidina calmodulina Proteínas reguladas por calmodulina heparina Fatores de coagulação, lipases, hormônios, receptores estoróides, etc Metais de transição Proteínas e peptídeos com resíduos de His expostos Sítios de ligação das proteínas A, G e L à imunoglobulina, que permitem a purificação de anticorpos por cromatografia de afinidade
aminoadenosine-3 ,5 -cyclic monophosphate. (ligante para proteínas com afinidade por cAMP ou cGMP) Ligante. grupo-específico. Especificidade. Proteína A. Região Fc de IgG. Proteína G. Concanavalina A. Grupos glicosil- ou manosil- Cibacron Blue. Várias enzimas, albumina. lisina. Plasminogênio, RNA ribossomal. arginina. proteinases tipo tripsina. benzamidina. calmodulina. Proteínas reguladas por calmodulina. heparina. Fatores de coagulação, lipases, hormônios, receptores estoróides, etc. Metais de transição. Proteínas e peptídeos com resíduos de His expostos. Sítios de ligação das proteínas A, G e L à imunoglobulina, que permitem a purificação de anticorpos por cromatografia de afinidade.")
33
Preparo da resina de afinidade:
Passo 1. Ativação da resina Passo 2. Acoplar o ligante Estratégias para acoplamento de ligantes à resina Reagente Alvo no ligante Para ligantes pequenos (Mr < 1.000) há o risco de impedimento estérico entre a matriz e a molécula-alvo, que causa dificuldade ou impede sua ligação à resina. A introdução de um braço espaçador diminue esse risco. Braço espaçador covalente entre a resina e o ligante Ligação reversível não covalente ligante Molécula-alvo (analito)
 há o risco de impedimento estérico entre a matriz e a molécula-alvo, que causa dificuldade ou impede sua ligação à resina. A introdução de um braço espaçador diminue esse risco. Braço espaçador covalente entre a resina e o ligante. Ligação reversível não covalente. ligante. Molécula-alvo (analito)")
34
Cromatografia de Partição
Princípio: Explora diferenças de solubilidade dos compostos em solventes com grau de hidrofobicidade diferentes, um polar e outro apolar. Aplicável especialmente á moléculas pequenas, como compostos orgânicos, aminoácidos, peptídeos, açúcares, lipídeos, etc. Apresenta limitações para uso com proteínas acima de 20-30kda, que são desnaturadas em presença de solvente orgânico. Fase estacionária (suporte sólido) X Fase móvel (líquido ou gás) Consiste de 2 sistemas: Papel ou placa de sílica Amostra na origem tempo t t tn direção do fluxo do solvente Compostos separados Cuba com solvente (fluxo por capilaridade) Suporte: PAPEL Fase Estacionária: Aquosa (H20 na celulose) Fase Móvel: Solvente orgânico (hidrofóbico) CROMATOGRAFIA EM PAPEL CROMATOGRAFIA DE FASE REVERSA Fase Estacionária: sílica (mineral apolar) Fase Móvel: Solvente aquoso ou água Suporte: SÍLICA (camada delgada ou TLC) distância de migração da substância distância de migração do solvente R f =
 X. Fase móvel. (líquido ou gás) Consiste de 2 sistemas: Papel. ou placa de sílica. Amostra na origem. tempo 0 t 1 t2 tn. direção do fluxo do solvente. Compostos separados. Cuba com solvente (fluxo por capilaridade) Suporte: PAPEL. Fase Estacionária: Aquosa (H20 na celulose) Fase Móvel: Solvente orgânico (hidrofóbico) CROMATOGRAFIA EM PAPEL. CROMATOGRAFIA DE FASE REVERSA. Fase Estacionária: sílica (mineral apolar) Fase Móvel: Solvente aquoso ou água. Suporte: SÍLICA (camada delgada ou TLC) distância de migração da substância. distância de migração do solvente. R. f. =")
35
HPLC: high performance (pressure) liquid chromatography
Inicialmente desenvolvida para cromatografia de fase reversa em coluna, hoje em dia abrange aplicações para todos os tipos de cromatografias. Característica diferencial da cromatografia convencional: partículas de resina com diâmetro muito pequeno (poucos microns) aumento da eficiência da separação em função do número maior de partículas de resina em um mesmo volume da coluna fase móvel - necessita bomba de alta pressão para ter fluxo através da coluna Desenho básico de um HPLC bombas Injetor de amostra Coluna e forno Coletor de frações Detector Controle e análise dos dados Duas bombas permitem eluição em gradiente Detector: índice de refração, absorbância UV ou Vis, fluorescência, etc. Coluna pode ser aquecida para melhorar a eluição diferencial na fase reversa
 aumento da eficiência da separação em função do número maior de partículas de resina em um mesmo volume da coluna. fase móvel - necessita bomba de alta pressão para ter fluxo através da coluna. Desenho básico de um HPLC. bombas. Injetor de amostra. Coluna e forno. Coletor de frações. Detector. Controle e análise dos dados. Duas bombas permitem eluição em gradiente. Detector: índice de refração, absorbância UV ou Vis, fluorescência, etc. Coluna pode ser aquecida para melhorar a eluição diferencial na fase reversa.")
36
Fase reversa: resinas de sílica derivatizadas com hidrocarbonetos de 2 C a 18 C
Absorbância a 214 nm Tempo ou volume de retenção % de acetonitrila 100 Moléculas não retidas Moléculas retidas Cromatograma de uma coluna de fase reversa, com eluição por um gradiente de acetonitrila Fase estacionária Função C18 –Si (CH3)2 C18 H37 C8 –Si (CH3)2 C8 H17 tC2 –Si C2 H5 Aminopropyl –Si (CH2)3 NH2 Cyanopropyl –Si (CH3)2 (CH2)3CN Diol –Si (CH2)3 OCH2CH(OH)CH2OH Condição de equilíbrio: em meio ácido (0.1% de ácido trifluoroacético) para aumentar hidrofobicidade (protonar as carboxilas) Eluição com gradiente crescente de solvente orgânico miscível com água - acetonitrila, metanol, propanol CN Fenil NH2 C4 C8 C18 Retenção na coluna: aumenta com o tamanho da cadeia
2 C18 H37. C8 –Si (CH3)2 C8 H17. tC2 –Si C2 H5. Aminopropyl –Si (CH2)3 NH2. Cyanopropyl –Si (CH3)2 (CH2)3CN. Diol –Si (CH2)3 OCH2CH(OH)CH2OH. Condição de equilíbrio: em meio ácido (0.1% de ácido trifluoroacético) para aumentar hidrofobicidade (protonar as carboxilas) Eluição com gradiente crescente de solvente orgânico miscível com água. - acetonitrila, metanol, propanol. CN Fenil NH2 C4 C8 C18. Retenção na coluna: aumenta com o tamanho da cadeia.")
37
A proteína pura é tratada com
aminoácido Para determinar a estrutura primária de uma proteína, é necessário primeiro conhecer a composição (número e tipos) de seus aminoácidos. Tempo de retenção (min) fluorescência A proteína pura é tratada com HCl 6N fervente para quebrar (hidrólise) as ligações peptídicas. A mistura resultante é submetida a métodos cromatográficos (fase reversa, troca iônica) para separar os diferentes aminoácidos. A posição do pico no cromatograma identifica o aminoácido. A área do pico quantifica o aminoácido.
 de seus aminoácidos. Tempo de retenção (min) fluorescência. A proteína pura é tratada com. HCl 6N fervente para quebrar (hidrólise) as ligações peptídicas. A mistura resultante é submetida a métodos cromatográficos (fase reversa, troca iônica) para separar os diferentes aminoácidos. A posição do pico no cromatograma identifica o aminoácido. A área do pico quantifica o aminoácido.")
38
Existem vários métodos para se determinar a sequência de aminoácidos de uma proteína. Aqui veremos como funcionam os dois métodos atualmente mais utilizados: Método de Edman: reação do aminoácido N-terminal da proteína com fenil-isotiocianato A proteína modificada é submetida a hidrólise ácida liberando o aminoácido N-terminal modificado, e este é identificado por cromatografia. Segue-se novo ciclo de reação com o próximo aminoácido na proteína, que se tornou o novo N-terminal. Espectrometria de massa: determinação das massas de fragmentos correspondendo a aminoácidos, retirados sequencialmente da proteína. Veremos mais sobre esse método na aula sobre eletroforese e proteômica.

39
ciclo 1 ciclo 2 hidrólise com ácido trifluoroacético hidrólise ácida vai para novo ciclo análise cromatográfica (troca iônica ou fase reversa) (1) (2) (3) acoplamento Método de Edman - sequenciamento de proteínas com PITC (fenil-isotiocianato) Cada ciclo de reação compreende 3 etapas: Reação da proteína com PITC, que se acopla ao grupo amino NH2- livre do aminoácido 1 (no exemplo, uma lisina, K); Hidrólise ácida da proteína conjugada com PITC libera a feniltiohidantoína (PTH) do aminoácido 1 e o restante da proteína, tornando o aminoácido 2 o novo resíduo N-terminal (no exemplo uma serina, S); Análise cromatrográfica do PTH-aminoácido e novo ciclo de reação com o novo N-terminal da proteína. Sequenciadores automatizados fazem todas as etapas, com capacidade para realizar 30 ciclos por dia, a partir de picomoles de proteína.
 (1) (2) (3) acoplamento. Método de Edman - sequenciamento de proteínas com PITC (fenil-isotiocianato) Cada ciclo de reação compreende 3 etapas: Reação da proteína com PITC, que se acopla ao grupo amino NH2- livre do aminoácido 1 (no exemplo, uma lisina, K); Hidrólise ácida da proteína conjugada com PITC libera a feniltiohidantoína (PTH) do aminoácido 1 e o restante da proteína, tornando o aminoácido 2 o novo resíduo N-terminal (no exemplo uma serina, S); Análise cromatrográfica do PTH-aminoácido e novo ciclo de reação com o novo N-terminal da proteína. Sequenciadores automatizados fazem todas as etapas, com capacidade para realizar 30 ciclos por dia, a partir de picomoles de proteína.")
40
Quando uma proteína possui mais de resíduos de aminoácidos, não é possível sequenciá-la diretamente pelo método de Edman. Proteína Total 150 a.a sequência obtida a partir da proteína intacta 30 aa Para obter a sequência completa de uma proteína, é necessário sequenciar vários peptídeos da mesma proteína, obtidos por diferentes tipos de quebra da cadeia, até haver sobreposição de suas sequências. Diferentes métodos são utilizados para obter-se diferentes peptídeos da proteína: A) enzimas proteolíticas, como tripsina (quebra em resíduos de Lys ou Arg) e quimotripsina (quebra em resíduos de Phe); B) tratamento com brometo de cianogênio (quebra em Met); e outros. proteína inteira peptídeos método A peptídeos método B
 enzimas proteolíticas, como tripsina (quebra em resíduos de Lys ou Arg) e quimotripsina (quebra em resíduos de Phe); B) tratamento com brometo de cianogênio (quebra em Met); e outros. proteína inteira. peptídeos método A. peptídeos método B.")
41
Sequenciamento de novo de proteínas por espectrometria de massas
Para sequenciar proteínas é preciso obter o espectro MS/MS de seus peptídeos. Isso significa 2 etapas de MS acopladas. Depois de fazer o MS da molécula inteira, esta é fragmentada dentro do aparelho por colisão com gases. Os fragmentos são então separados e analisados por MS. hélio ou argônio A ligação peptídica se quebra formando fragmentos típicos, como os íons b e y mostrados na figura acima, que terão massas diferentes de acordo com o radical R de cada aminoácido.

42
Espectro MS/MS do peptídeo tríptico GLSDGEWQQVLNVWGK.
AA Codes Mono. AA Codes Gly G Asp D Ala A Gln Q Ser S Lys K Pro P Glu E Val V Met M Thr T His H Cys C Phe F Leu L Arg R Ile I CMC Asn N Tyr Y - Trp W Espectro MS/MS do peptídeo tríptico GLSDGEWQQVLNVWGK. Analiza-se o espectro buscando diferenças de m/z equivalentes a um resíduo de aminoácido, que permitem conhecer a seqüência do peptídeos

43
Síntese de peptídeos Quim. Nova, Vol. 27, No. 5, 781-789, 2004 H2N
bloq COOH bloq DNA recombinante Química: uso de reagente químico para ativar o ácido carboxílico de um Nα-acil-aminoácido, o qual sofre o ataque nucleofílico do grupo α- amino de outro aminoácido - indicado para sequências de até 20 aa. - estereo-isômeros, purificação dos produtos Biocatálise: reversão da proteólise - diminuição da atividade da água no meio reacional reverte a ação de enzimas proteolíticas - termolisina, pepsina, subtilisina, tripsina, etc - vantagens:, não forma misturas racêmicas e sub-produtos, condições brandas

44
Síntese de peptídeos Fmoc (9-fluorenilmetoxicarbonila) - protetor do grupo α- amino dos doadores de acila, estável ao ácido trifluoroacético (TFA) e lábil a bases orgânicas – todas as outras carboxilas são protegidas por reagentes lábeis ao TFA e estáveis a bases orgânicas Boc (t-butiloxicarbonila) - protetor do grupo α- amino dos doadores de acila lábil ao (TFA) – todas as outras carboxilas são protegidas por reagentes estáveis a este ácido e lábeis a ácidos inorgânicos fortes ou hidrogenólise.
 - protetor do grupo α- amino dos doadores de acila, estável ao ácido trifluoroacético (TFA) e lábil a bases orgânicas – todas as outras carboxilas são protegidas por reagentes lábeis ao TFA e estáveis a bases orgânicas. Boc (t-butiloxicarbonila) - protetor do grupo α- amino dos doadores de acila lábil ao (TFA) – todas as outras carboxilas são protegidas por reagentes estáveis a este ácido e lábeis a ácidos inorgânicos fortes ou hidrogenólise.")
45
Síntese de peptídeos Robert B. Merrifield – Prêmio Nobel em Química em 1984

46
Síntese de peptídeos

47
ELETROFORESE ELETROFORESE + D T SUPORTE X pH MEIO 1. pH / tampão
CONDIÇÕES QUE DETERMINAM A SEPARAÇÃO SUPORTE X pH MEIO + D T 1. pH / tampão 2. Suporte - papel : corrente alta (calor) uso para peptídeos e aminoácidos - poliacrilamida : proteínas ac. nucleicos não desnaturante ou nativa desnaturante e redutor peso molecular composição de subunidades focalização isoelétrica: PI bidimensional - agarose : ácidos nucléicos Imunoeletroforese Proteínas nativas
 uso para peptídeos e aminoácidos. - poliacrilamida. : proteínas. ac. nucleicos. não. desnaturante ou nativa. desnaturante. e redutor. peso molecular. composição de subunidades. focalização isoelétrica: PI. bidimensional. - agarose. : ácidos nucléicos. Imunoeletroforese. Proteínas nativas.")
48
_ + Reação de eletrólise da água Uso de tampão concentrado
Uso de indicador de pH como marcador de corrida: azul de bromofenol

49
Eletroforese em Gel de Poliacrilamida (PAGE)
polyacrylamide persulfate + 7 a 15% [acrilamida] – maioria das proteínas [ ] mais baixas – gel muito mole – suporte com agarose Gradiente de acrilamida – aumenta poder de resolução cuba vertical para mini-gel (10 X 8 cm)
")
50
Proteína Ponto Isoelétrico Pepsina < 1.0 Ovalbumina (galinha) 4.6 Albumina Sérica (humana) 4.9 Tropomiosina 5.1 Insulina (bovina) 5.4 Fibrinogênio (humano) 5.8 g - Globulina 6.6 Colágeno Mioglobina (cavalo) 7.0 Hemoglobina (humana) 7.1 Ribonuclease A (bovina) 7.8 Citocromo c 10.6 Histona (bovina) 10.8 Lisozima (galinha) 11.0 Salmina ( salmon ) 12.1 Separação na eletroforese nativa é influenciada pela carga, massa e forma da molécula 18.000 64.000
 5.4. Fibrinogênio (humano) 5.8. g. - Globulina Colágeno. Mioglobina. (cavalo) 7.0. Hemoglobina (humana) 7.1. Ribonuclease. A (bovina) 7.8. Citocromo. c Histona (bovina) Lisozima (galinha) Salmina. ( salmon. ) Separação na eletroforese nativa é influenciada pela carga, massa e forma da molécula")
51
A B C Efeitos do SDS desnaturação uniformiza a forma das proteínas;
SDS (dodecil sulfato de sódio + 2-mercaptoetanol Efeitos do SDS desnaturação uniformiza a forma das proteínas; mascara a carga natural das proteínas no pH da corrida, fazendo com que todas moléculas migrem para o anôdo; facilita o efeito de redutores

52
Salmonella tiphymurium
SDS-PAGE Salmonella tiphymurium

53
Determinação da massa molecular de proteínas
Curva de calibração de SDS-PAGE a 15% Mobilidade relativa (Rf) Massa molecular (kD) Mobilidade relativa = distância percorrida pela banda X distância percorrida pelo marcador da corrida 97.4 87.0 45.0 29.0 21.0 12.5 6.5 (-) (+)
 Massa molecular (kD) Mobilidade relativa = distância percorrida pela banda X. distância percorrida pelo marcador da corrida (-) (+)")
54
_ Eletroforese – focalização isoelétrica + Aplicações:
• migração através de um gradiente de pH • proteínas “focalizam” nos seus P.I. Anfólitos: Misturas de compostos poliaminados/policarboxilados (patenteados) Gel não tamponado polimerizado com anfólitos _ 1ª corrida: anfólitos criam gradiente 2ª corrida: amostras são aplicadas 3.0 Após a corrida: pedaços do gel são analisados quanto ao seu pH. 4.0 Aplicações: Determinação do PI Isoformas da mesma proteína: glicosilação fosforilação mutações pontuais pH 5.0 6.0 pH + Marcadores de P.I. conhecidos
 Gel não tamponado polimerizado com anfólitos. _. 1ª corrida: anfólitos criam gradiente. 2ª corrida: amostras são aplicadas Após a corrida: pedaços do gel são analisados quanto ao seu pH Aplicações: Determinação do PI. Isoformas da mesma proteína: glicosilação. fosforilação. mutações pontuais. pH pH. + Marcadores de P.I. conhecidos.")
55
Eletroforese Bidimensional
- P I + 3 3.5 4 4.5 5 5.5 Origem 2 6 7 8 9 10 PM x 103 1a.dimensão: focalização isoelétrica 2a.dimensão: SDS-PAGE (perpendicular a 1a.)
")
56
Proteínas com mesmo pI Proteínas com a mesma massa PROTEOMA: análise simultânea de até proteínas permite comparação de todas as proteínas expressas por uma célula em dois momentos diferentes: análise do efeito de hormônios, análise do efeito de drogas; estados fisiológicos (desenvolvimento); condições patológicas diversas (vírus, bactérias,etc.) Eletroforese 2D de proteínas totais (proteoma) de Escherichia coli
; condições patológicas diversas (vírus, bactérias,etc.) Eletroforese 2D de proteínas totais (proteoma) de Escherichia coli.")
57
Gel nativo: sem fervura das amostras pode ter SDS no gel de separação
Conformação nativa, oligomerização Zimogramas: estudos de enzimas, afinidade Gel copolimerizado (gelatina, amido, etc) Gel overlay (gel de agarose) Proteínas nativas ou renaturadas no gel por remoção lenta do SDS por troca com detergente não iônico
 Gel overlay (gel de agarose) Proteínas nativas ou renaturadas no gel por remoção lenta do SDS por troca com detergente não iônico.")
58
Antígeno B do Echinococcus granulosus
Rhipicephalus microplus Cys-endopeptidase Multímeros de subunidades de 8 kDa Estrela et al, 2010 Comp. Biochem. Physiol 15% SDS-PAGE Monteiro et al., 2011 PLoS Neglected Tropical Diseases 12% SDS-PAGE- copolimerizado com 0.1 % hemoglobina - após corrida, retirada do SDS troca por Triton X-100 (3h) - incubação a pH 4,0, 18h/37oC. Superoxide dismutases from Metarhizium. anisopliae Urease de Canavalia ensiformis (hexâmero de 90 kDa) Ni2+ Cu2+ Zn2+ Hg2+ 3% PAGE 8.5% non-denaturing gels - atividade das SODs revelada Inibição da redução do NBT em presença de TEMED e riboflavina Efeito de Cu2+ - precipitação Follmer & Carlini, 2005 Arch. Biochem. Biophys. Tolfo Bittencourt et al., Res. Microbiol
 - incubação a pH 4,0, 18h/37oC. Superoxide dismutases from Metarhizium. anisopliae. Urease de Canavalia ensiformis. (hexâmero de 90 kDa) Ni2+ Cu2+ Zn2+ Hg2+ 3% PAGE. 8.5% non-denaturing gels. - atividade das SODs revelada Inibição da redução do NBT em presença de TEMED e riboflavina. Efeito de Cu2+ - precipitação. Follmer & Carlini, Arch. Biochem. Biophys. Tolfo Bittencourt et al., 2004 Res. Microbiol.")
59
Métodos imunoquímicos aplicados a proteínas
estrutura e propriedades de imunoglobulinas anticorpos policlonais e monoclonais: produção e purificação imunodiagnóstico: aglutinação X lise imunoprecipitação: difusão em gel, imunoeletroforese ELISA, Western blot imunohistoquímica e imunofluorescência

60
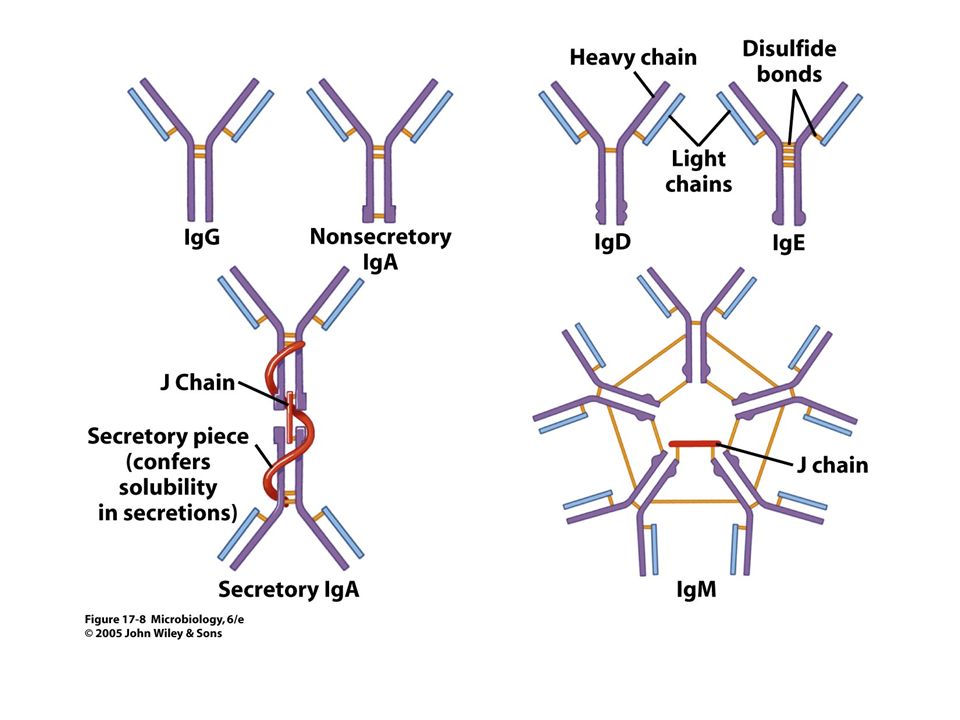
Anticorpos são imunoglobulinas.
Função Estrutura Cadeia H Cadeia L Imunoglobulina classe Sub-classe g1 k ou l IgG IgG1 g2 IgG2 IgG3 g4 IgG4 a1 IgA IgA1 a2 IgA2 m IgM d IgD e IgE Cadeia H (pesada) – d Cadeia L (leve) – d anticorpo antígeno Complexo antígeno-anticorpo Porção N-terminal das cadeias L e H ~ 120 aminoácidos variáveis restante da cadeia é constante
 – d. Cadeia L (leve) – d. anticorpo. antígeno. Complexo antígeno-anticorpo. Porção N-terminal das cadeias L e H. ~ 120 aminoácidos variáveis. restante da cadeia é constante.")
62
Mecanismos através dos quais os anticorpos executam as funções de defesa:
Neutralização (Ag solúveis) Opsonização (Ag particulados) Imunecomplexo (todos Ags)
 Opsonização (Ag particulados) Imunecomplexo (todos Ags)")
63
Propagação clonal de linfócitos B ou plasmócitos
Produção de Anticorpos: imunização ativa Propagação clonal de linfócitos B ou plasmócitos Um linfócito B sempre produzirá imunoglobulinas para o mesmo antígeno, mas poderá variar a classe de anticorpos produzidos

64
Anticorpos policlonais versus Anticorpos monoclonais
Há vários determinante antigênico ou epitopo em uma proteína - tamanho: 5 a 6 aminoácidos - podem ser sequenciais ou conformacionais

65
Produção de Anticorpos Monoclonais
Reconhecimento de apenas um determinante antigênico tipo único de imunoglobulina Vantagens e desvantagens

66
Imunoprecipitação Complexos Ag-Ac insolúveis - Precipitado é máximo na
Zona de equivalência - Excesso de Ag ou de Ac Fazem complexos solúveis

67
Imunoprecipitação em gel de agarose

68
Imunodifusão dupla Permite comparar antígenos e detectar determinantes antigênicos em comum Identidade total Identidade parcial Ausência de identidade

69
Proteínas bacterianas com afinidade por Imunoglobulinas
- Servem como ligantes em matrix de afinidade Servem como 2º.ligantes em testes imunoenzimáticos Recombinant Protein L Native Protein A Recombinant Protein A Recombinant Protein G Protein A/G Source Peptostreptococci Staphylococcus aureus Bacillus Streptococci Molecular Weight 35,800 42,000 44,600 22,000 50,449 Number of Binding Sites for IgG 4 5 2 Albumin Binding Site no Optimal Binding pH 7.5 8.2 5-8.2 Binds to VLκ Fc bead

70
Imunobeads: adsorção complexo Ag:Ac

71
Ensaios Imunoenzimáticos ou ELISA
cor + 1º.Ac Ag S E ELISA sandwich + 2º.Ac S cor Ac 2º. Ac 1º. Ag placa Abs (intensidade da cor) Concentração do Ag ou Ac Padronização do ELISA
 Concentração do Ag ou Ac. Padronização do ELISA.")
72
ELISA Competitivo é adequado para identificação e quantificação tanto do antígeno como do anticorpo.
Para a determinação de Ag, o Ag presente na amostra compete com Ag marcado com uma enzima, para se ligar ao Ac imobilizado. A cor desenvolvida na revelação é indiretamente proporcional à concentração de Ag na amostra. O Radioimunoensaio (RIA) baseia-se no mesmo princípio, utilizando Ag marcado com um radioisótopo para competir com o Ag frio presente na amostra. (1) (2) Variante para pesquisa do Ac - Quanto maior a concentração do antígeno a ser medido, maior será o deslocamento do antígeno marcado, permitindo a quantificação.
 baseia-se no mesmo princípio, utilizando Ag marcado com um radioisótopo para competir com o Ag frio presente na amostra. (1) (2) Variante para pesquisa do Ac. - Quanto maior a concentração do antígeno a ser medido, maior será o deslocamento do antígeno marcado, permitindo a quantificação.")
73
Western blot Anticorpo secundário marcado com: - enzima - radioativo
- fluorescente

74
Imunofluorescência: anticorpos marcados com diferentes fluoróforos
vermelho (rodamina), verde (fluoresceína) GluR1 clusters in hippocampal cultured neurons MAP2-(neurons) and GFAP-(astrocytes) positive cells in hippocampal cell culture Presynaptic terminals showned by immunostainig of specifically presynaptic protein Synapsin in cultured hippocampal neuron
, verde (fluoresceína) GluR1 clusters in hippocampal cultured neurons. MAP2-(neurons) and GFAP-(astrocytes) positive cells in hippocampal cell culture. Presynaptic terminals showned by immunostainig of specifically presynaptic protein Synapsin in cultured hippocampal neuron.")
Apresentações semelhantes