Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Comissão de Fibrose Cística

2
Nature July 2009; 460/9 Figura 1: Lap-Chee Tsui, Francis Collins e Jack Riordan (esquerda para a direita) celebram a descoberta, em 1989, do gene CFTR com uma paciente fibrocística. . Figura 2: Vinte anos após a descoberta do gene CFTR.
 celebram a descoberta, em 1989, do gene CFTR com uma paciente fibrocística. . Figura 2: Vinte anos após a descoberta do gene CFTR.")
3
Alt. Splicing 3849+10kbC–>T
Mutações no gene CFTR. Normal I II III IV V Nonsense G542X Frameshift 394delTT Missense Missense G551D Missense R117H Alt. Splicing kbC–>T AA deletion DF508 Missense A455E No synthesis Block in processing Altered conductance Block in regulation Reduced synthesis There are a variety of mechanisms by which mutations in CFTR lead to quantitative or qualitative reductions in its function in affected individuals. Julian Zielenski and Lap Chee Tsui proposed 5 general functional classes of CF mutations. In class I mutations, there is essentially no message synthesis. These are usually the result of premature stop codons (ex. G542X) or frameshift mutations. Mutations in class I usually produce few or no functioning CFTR chloride channels. In class II mutations, there is typically a full length message made but a block in processing occurs. In the most common CF mutation, F508, there is a deletion of an amino acid, phenylalanine. This mutant CFTR is degradaded in the endoplasmic reticulum and fails to reach the apical surface membrane. Class III mutations have defective regulation of CFTR, even though the mutant protein reaches the cell surface. In class IV mutations, CFTR reaches the apical surface membrane, but conduction is defective due to altered conduction or gating channel properties. Class V mutations are associated with reduced synthesis. However, the protein present has normal activity. These mutations often lead to variable clinical presentations as demonstrated in the next slide. Tsui LC 2000
 or frameshift mutations. Mutations in class I usually produce few or no functioning CFTR chloride channels. In class II mutations, there is typically a full length message made but a block in processing occurs. In the most common CF mutation, F508, there is a deletion of an amino acid, phenylalanine. This mutant CFTR is degradaded in the endoplasmic reticulum and fails to reach the apical surface membrane. Class III mutations have defective regulation of CFTR, even though the mutant protein reaches the cell surface. In class IV mutations, CFTR reaches the apical surface membrane, but conduction is defective due to altered conduction or gating channel properties. Class V mutations are associated with reduced synthesis. However, the protein present has normal activity. These mutations often lead to variable clinical presentations as demonstrated in the next slide. Tsui LC")
6
Comissão de Fibrose Cística da SBPT
Auxiliar a SBPT nos assuntos referentes a Fibrose Cística. Divulgação, educação continuada, participação nos congressos Integração com outras sociedades e associações: (nacionais) GBEFC Sociedade Brasileira de Pediatria, Sociedade Brasileira Genética, Sociedade Brasileira de Microbiologia, Associação Brasileira de Assistência a Mucoviscidose (ABRAM) 4. Construir a historia da FC no Brasil.
 GBEFC. Sociedade Brasileira de Pediatria, Sociedade Brasileira Genética, Sociedade Brasileira de Microbiologia, Associação Brasileira de Assistência a Mucoviscidose (ABRAM) 4. Construir a historia da FC no Brasil.")
7
Contatos: European Cystic Fibrosis Society Cystic Fibrosis Foundation

8
A Fibrose Cística é uma DPOC A FC é uma Discinesia ciliar secundaria
Qual o melhor manejo pelo Pneumologista?

9
Comissão de Fibrose Cística da SBPT
5. Estimular a elaboração de um protocolo de assistência a os pacientes com FC. 6. Estimular o desenvolvimento de pesquisas e publicações no Jornal Brasileiro de Pneumologia . 7. Divulgar teses e publicações brasileiras sobre FC no site da Comissão 8. Estimular o diagnóstico da FC no Brasil. 9. Estimular a transição de pacientes adolescentes e adultos que estão sendo atendidos nos centros pediátricos para os centros de atendimento interdisciplinares de adultos.

10
Ações DIANOSTICO P Ped P Adulto Recursos tratamentos Qual o papel do Pneumologista na equipe interdisciplinar?

12
Outros profissionais como sócios para a SBPT
Pesquisas multicêntricas Potencial nasal Biópsia retal Cursos de capacitação

13
A Fibrose Cística no Brasil: Problemas
Elevada proporção de casos não diagnosticados Diagnóstico tardio Poucos serviços em relação à demanda real e à estimada Reduzido número de profissionais de saúde envolvidos Doença subestimada pelas autoridades de saúde (baixos investimentos e reduzidas verbas de custeio) Produção científica ainda tímida
 Produção científica ainda tímida.")
14
Comissão de Fibrose Cística da SBPT
Solicitação do Dr. Renato Maciel Presidente do Congresso SBPT 2012 Duas indicações de palestrantes internacionais da sua área como indicação para o Congresso SBPT 2012 em Belo Horizonte. Tais indicações deverão ter seu mérito esclarecido.

15
Margarida Amaral Fala português
Reconhecida no mundo todo como uma das maiores pesquisadoras em FC Conferencista do Congresso Americano de FC. (Homenageada) Conferencista do Congresso Europeu de FC. (Homenageada) Conferencista do Congresso brasileiro de FC. Numerosos trabalhos em periódicos ISI e Medline
 Conferencista do Congresso Europeu de FC. (Homenageada) Conferencista do Congresso brasileiro de FC. Numerosos trabalhos em periódicos ISI e Medline.")
16
Margarida Amaral e-mail: mdamaral@fc.ul.pt
Tel: (ext ) Fax:
 Fax:")
17
Felix Ratgen Dr. Felix Ratjen, Professor of Pediatrics, University of Toronto Division Chief, Respiratory Medicine H.E. Seller's Chair in Cystic Fibrosis The Hospital for Sick Children 555 University Avenue , Toronto, Ontario, Canada, M5G 1X8

18
Muito grato pela parceria de sempre, Abraços,
Prezados, Venho solicitar que cada comissão e departamento da SBPT elabore um caso clínico de sua respectiva área a fim de que seja divulgado no nosso site. Precisamos dinamizar este importante setor da home page, responsável por grande número de acessos atualmente. Para tal, definimos como data limite para a entrega do material como sendo o dia da reunião da diretoria e comissões em SP, ou seja 11/12/11. Sugerimos que se visite os já existentes na página para devida padronização do formato de apresentação. Muito grato pela parceria de sempre, Abraços, Dr. Bernardo Maranhão Diretor Científico . SEPS 714/914 - Bloco E - Sala 220/223. Asa Sul – Brasília/DF Fone/fax:

19
8 sugestões de aulas e respectivas indicações de palestrantes até o dia 23/12, a fim de que possamos organizar as grades do Curso Nacional de Atualização e PECs. Dr . Bernardo Maranhão Diretor Científico SBPT Sugestões de aulas: 1) Fibrose Cística no Adulto. 2) Fibrose Cística na Criança. 3) Transição Criança adolescente adulto. Quando Como e porque? 4) Fatores de piora da função pulmonar na Fibrose Cística 5) Manejo de germes multiressistentes na Fibrose Cística 6) Diagnóstico da Fibrose Cística 7) Exacerbação da doença pulmonar na Fibrose Cística 8) Medicamentos para o manejo pulmonar da Fibrose cística
 Fibrose Cística no Adulto. 2) Fibrose Cística na Criança. 3) Transição Criança adolescente adulto. Quando Como e porque 4) Fatores de piora da função pulmonar na Fibrose Cística. 5) Manejo de germes multiressistentes na Fibrose Cística. 6) Diagnóstico da Fibrose Cística. 7) Exacerbação da doença pulmonar na Fibrose Cística. 8) Medicamentos para o manejo pulmonar da Fibrose cística.")
20
Caso clínico SRMP, 30 anos, professora, branca, grávida primigesta,
QP: Tosse noturna discreta nos últimos 3 anos. Sempre foi saudável. Nunca teve pneumonia, nunca internou, não é tabagista, nega asma, nega dispnéia. IC: nega outras queixas Exame físico: BEG, nutrida (IMC=26), afebril, anictérica, acianótica. Coração e pulmões: ndn
, afebril, anictérica, acianótica. Coração e pulmões: ndn.")
21
Caso clínico Pedido pessoal para diagnóstico pré-natal de aberrações cromossômicas e análise da mutação das doenças metabólicas incluindo CF Resultado: duas mutações CFTR detectado: DF 508 / R117H diagnóstico materno: FC Mãe desgastada, consulta quanto à interrupção da gravidez Qual seria a sua abordagem?

22
VEF1: 95% CVF: 95% VEF/CFV: 100% FEF25-75: 85% Espirometria
Tomografia computadorizada de Tórax VEF1: 95% CVF: 95% VEF/CFV: 100% FEF25-75: 85%

23
Triagem neonatal (teste do pezinho) 4. Quadro clínico
Fibrose Cística O que é FC? Fisiopatologia Diagnóstico Triagem neonatal (teste do pezinho) 4. Quadro clínico Rosestein BJ et al. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998
 4. Quadro clínico. Rosestein BJ et al. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr")
24
É uma doença genética. Não existe tratamento.
FC resulta de aproximadamente mutações no gene regulador da proteína condutora de cloro (CFTR) através da parte apical das células de vários órgãos. (Cromossoma 7) É uma doença genética. Não existe tratamento.
 através da parte apical das células de vários órgãos. (Cromossoma 7) É uma doença genética. Não existe tratamento.")
26
Co-descobridores do gene CFTR
The discovery of the CF gene was the culmination of the efforts of 1000s of scientists, caregivers, patients and their families. But, it was the vision and creativity of these three men that allowed us to cross the finish line. Fortunately for the field of CF, all three men are still involved in CF. Lap-Chee has been involved in the characterization of the more than 1,000 CF gene mutations and is leading the effort in finding how genes may modify the phenotype in CF. Jack Riordan, now at the Mayo Clinic in Scottsdale, Arizona, is continuing his efforts to characterize CFTR and its expression. Francis Collins as the Institute Director of the National Human Genome Research Institute and quarterback of the Human Genome Project has helped to usher in the era of Genomic Medicine -- and it is this era that will provide the greatest hope for human health in the decades ahead. Lap-Chee Tsui, M.D. John Riordan, Ph.D. Francis S. Collins, M.D., Ph.D.

27
2) Fisiopatologia Fibrose Cística
Rosestein BJ et al. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998

28
Doença Pulmonar na Fibrose Cística (FC)
Defeito no gene da FC CFTR Defeituosa/Deficiente/Ausente Anormalidades no líquido de superficie das VA Obstrução das VA Inflamação Infecção Bronquiectasias Konstan, Davis 2001

29
Alterações pulomonares precoces na FC patofisiologia
Ratjen und Döring, Lancet 2003 a b c d e f

30
Neutrophil Inflammation in CF
LTB4, IL-8 PMN Failure of opsonophagocytosis Bacterial persistence Epithelial IL-8 Secretion Elastin degradation IgG cleavage CR1, C3bi cleavage Elastase O2 H2O2 . Plugging of airways DNA Structural damage Bronchiectasis Konstan 2001

31
Mucociliary Clearance in CF
Regnis, et. al. Am J Resp Crit Care Med 1994

32
MEF50 MEF75-25 FEV1 FVC Gravidade
Os Parâmetros de Sensiblidade da Função Pulmonar Variam com a gravidade da Doença Inicial Avançada Estágio final % função pulmonar 100 MEF50 MEF75-25 FEV1 FVC Menos Mais Gravidade Tiddens, Pediatr Pulmonol 2002

33
Quando pensar em FC? Bronquiecatasias

34
Lactentes com bronquiolite Obliterante ou sibilancia grave
Quando pensar em FC? Lactentes com bronquiolite Obliterante ou sibilancia grave

35
Ileo meconial

36
Inflamatometria Inflamatólogos Inflamatófilos Inflamatófobos
Serviço de Pneumologia Pediatrica - Depto. Pediatria - FCM/HC - UNICAMP.

38
Quando pensar em FC? Causas de desnutrição na FC
Mal absorção intestinal Diminuição de enzimas pancreáticas Deficiência de NaHCO3 Anomalias de sais biliares Alterações na motilidade Baixa ingesta de calorias Infecções repetidas

39
Sinfain M et al. Diagnosing Cystic Fibrosis at All Ages.
(Clin Pulm Med 2010;17: 14–19) Doença do trato respiratório superior Polipose nasal e sinusite crônica Sakano E et al. International Journal of Pediat. Otorhinolaryngol. (2007) 71, 41-50 Doença do trato respiratório inferior BVA grave Pneumonia ou bronquite recorrente Asma de difícil controle Atelectasia crônica Atelectasia crônica com infecção por micob não tuberculosas Bronquiectasias inexplicável Tosse crônica produtiva Hipocratismo digital Infecção pulmonar por bactérias Gram – ou S. aureus Hemoptise
 Doença do trato respiratório superior Polipose nasal e sinusite crônica Sakano E et al. International Journal of Pediat. Otorhinolaryngol. (2007) 71, Doença do trato respiratório inferior. BVA grave Pneumonia ou bronquite recorrente Asma de difícil controle Atelectasia crônica Atelectasia crônica com infecção por micob não tuberculosas Bronquiectasias inexplicável Tosse crônica produtiva Hipocratismo digital Infecção pulmonar por bactérias Gram – ou S. aureus Hemoptise.")
40
Sinfain M et al. Diagnosing Cystic Fibrosis at All Ages.
(Clin Pulm Med 2010;17: 14–19) Doença gastrointestinal Íleo meconial ou síndrome de plug de mecônio Icterícia neonatal prolongada Prolapso retal Intussuscepção Recorrente Insuficiência hepática inexplicada Pancreatite crônica ou recorrente Anormalidades nutricionais Falha de crescimento Esteatorréia Hipoproteinemia Deficiências de vitamina solúvel em gordura Coagulopatia (vitamina K) Anemia hemolítica (vitamina E
 Doença gastrointestinal Íleo meconial ou síndrome de plug de mecônio Icterícia neonatal prolongada Prolapso retal Intussuscepção Recorrente Insuficiência hepática inexplicada Pancreatite crônica ou recorrente. Anormalidades nutricionais Falha de crescimento Esteatorréia Hipoproteinemia Deficiências de vitamina solúvel em gordura Coagulopatia (vitamina K) Anemia hemolítica (vitamina E.")
41
Sinfain M et al. Diagnosing Cystic Fibrosis at All Ages.
(Clin Pulm Med 2010;17: 14–19) Outras condições: Desidratação hiponatrêmica Alcalose metabólica Gosto salgado da pele Azoospermia Criptorquidismo Ausencia do ducto deferente
 Outras condições: Desidratação hiponatrêmica Alcalose metabólica Gosto salgado da pele Azoospermia Criptorquidismo Ausencia do ducto deferente.")
42
3) Diagnóstico Fibrose Cística
Rosestein BJ et al. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998

43
FC: diagnóstico Rosestein BJ et al. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998 Características fenotípicas (1 ou mais): . Doença sinusal e/ou pulmonar crônica . Alterações gastrointestinais ou nutricionais . Síndrome da perda de sal . Anormalidades urogenitais resultando em azoospermia obstrutiva OU Cloro no suor > 60 mEq/L em 2 dosagens Histórico de irmão com FC Identificação de 2 mutações do gene CFTR Teste de triagem neonatal positivo Demonstração de alteração do transporte iônico no epitelio nasal Ausência do canal de cloro em epitélio intestinal Rosestein BJ et al. J Ped. 1998, 132: Para o diagnóstico é necessário pelo menos 1 dos itens da coluna A e 1 da coluna B.
: . Doença sinusal e/ou pulmonar crônica. . Alterações gastrointestinais ou nutricionais. . Síndrome da perda de sal. . Anormalidades urogenitais resultando em azoospermia obstrutiva. OU. Cloro no suor > 60 mEq/L em 2 dosagens. Histórico de irmão com FC. Identificação de 2 mutações do gene CFTR. Teste de triagem neonatal positivo. Demonstração de alteração do transporte iônico no epitelio nasal. Ausência do canal de cloro em epitélio intestinal. Rosestein BJ et al. J Ped. 1998, 132: Para o diagnóstico é necessário pelo menos 1 dos itens da coluna A e 1 da coluna B.")
44
O diagnóstico de fibrose cística
1) Quadro clinico compatível e cloro no suor >60 mmol /L ou 2) Valores de cloro está na faixa intermediária: (30-59 mmol / L para crianças < 6 meses de idade, (40-59 mmol / L para os indivíduos mais velhos) e apresenta duas mutações do gene CFTR identificadas Farrell PM, et al. Guidelines for diagnosis of cystic fi brosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr 2008; 153: S4–14
 Quadro clinico compatível e cloro no suor >60 mmol /L. ou. 2) Valores de cloro está na faixa intermediária: (30-59 mmol / L para crianças < 6 meses de idade, (40-59 mmol / L para os indivíduos mais velhos) e apresenta duas mutações do gene CFTR identificadas. Farrell PM, et al. Guidelines for diagnosis of cystic fi brosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J Pediatr 2008; 153: S4–14.")
45
Teste do Tripsinogênio Imunorreativo: ITR ou TIR
Precursor da tripsina Está aumentado no sangue dos pacientes com FC Sensibilidade 95% e especificidade 30-75% Dosar até o terceiro dia e entre a segunda e a quarta semana. Corte: 70mg/ml Rosestein BJ et al. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998

46
Algoritimo para o screening neonatal para FC
Amostra de sangue TIR Resultado < cut-off Resultado cut-off Repete a TIR cut-off Cloro no suor e/ou Estudo genetico com 1 ou 2 mutações <cut-off Screening negativo Essentially a neonatal screening program is performed by measuring immunoreactive trypsin in a dried blood sample similar to what is done in the Guthrie test. Samples exceeding a certain cut off level are processed and this may differ among centers. Ie some go directly into a DNA analysis other retest the child. A further difference among centers is the pannle of mutations which are looked for. In some cases only the prevalent in other it differs according to the local epidemiology of the mutations The screening program is not designed to catch all cases but to analyse the portion of the population with the highest probability to have (severe) CF. A number of patients will be lost: those with borderline IRT levels and those with two rare mutations. Diagnostico confirmado Encaminhar ao Centro de referencia
 CF. A number of patients will be lost: those with borderline IRT levels and those with two rare mutations. Diagnostico confirmado. Encaminhar ao Centro de referencia.")
47
Triagem Neonatal para Fibrose Cística
Os RN triados para a FC podem se beneficiar com o diagnóstico e tratamento precoces. Melhorar o crescimento Melhorar a função pulmonar Reduzir internações hospitalares Acrescentar anos à vida.

48
Triagem Neonatal (TNN) para FC
Embora a TNN positiva não seja um teste diagnóstico definitivo para a FC ela pode levar a testes que permitam excluir ou confirmar um diagnóstico de FC. A CFF, CDC, GBEFC, ECFS, recomendam a triagem neonatal para FC

49
TNN para FC e seu impacto na prática clínica diária:
Dúvidas, sofrimento, insegurança para os pais, falso-positivos, etc. Fluxo da TNN Onde realizar o teste do suor e como encaminhar os positivos para o centro de referência? Orientação sobre sintomas clínicos da FC Qual o impacto do diagnóstico tardio. Rosestein BJ et al. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998

50
CF CENTER VERONA – DIAGNOSIS 1958-2000
In this area neonatal screening programs have been implemented since the seventies and progressively all the neonates have been screened In the last 6-8 years most of our diagnosis in children resulted from neonatal screening

51
CENTER FOR CYSTIC FIBROSIS VERONA DIAGNOSIS OF CYSTIC FIBROSIS

52
Desafios no diagnóstico Fibrose Cística
Estado do Paraná de 2001 a 2004: IRT (TIR) realizadas: Positivas: (0,9%) Segundo IRT: positivo em bebes: Positivo em 478: (12,5%) Aproximadamente: 1:100 RN terá a primeira IRT positiva. 1:10 RN terá a segunda IRT positiva De 10 pacientes com a segunda IRT alterada: 1 FC para cada nascidos vivos Santos GP et al. Jornal de Pediatria 2005, 81: 240: -4
 realizadas: Positivas: (0,9%) Segundo IRT: positivo em bebes: Positivo em 478: (12,5%) Aproximadamente: 1:100 RN terá a primeira IRT positiva. 1:10 RN terá a segunda IRT positiva. De 10 pacientes com a segunda IRT alterada: 1 FC para cada nascidos vivos. Santos GP et al. Jornal de Pediatria 2005, 81: 240: -4.")
53
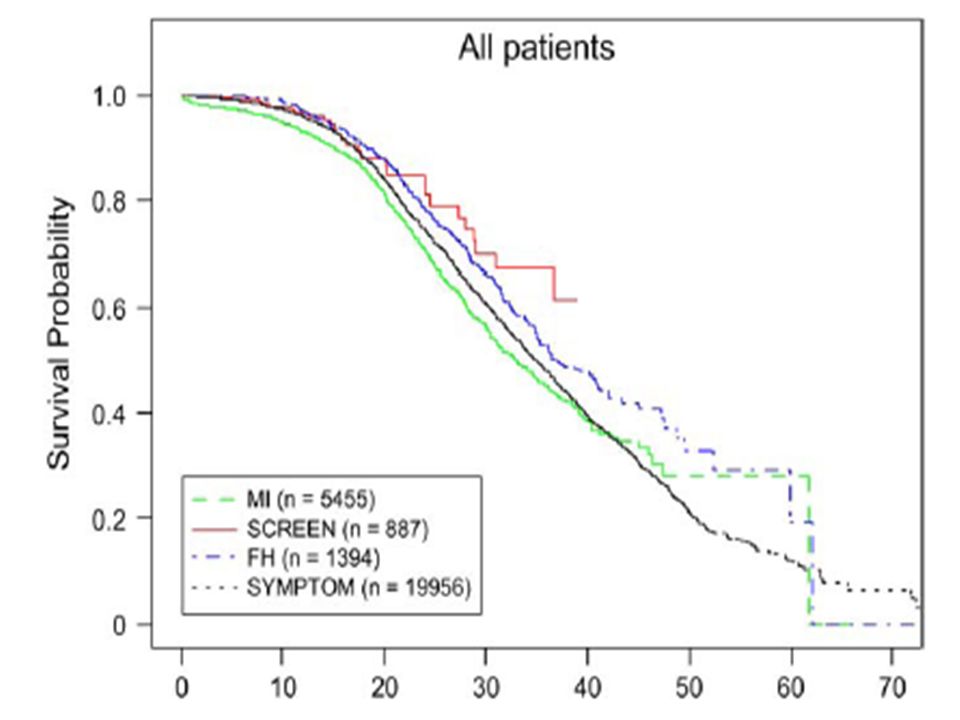
27.692 pacientes com FC divididos em 4 grupos:
Lai Hj et al. Vantagens para a sobrevida de pacientes com FC diagnosticados pela Triagem Neonatal. Evidências da CFF dos EUA. J Pediatr 2005;147:S57-S63) pacientes com FC divididos em 4 grupos: Diagnóstico prenatal ou neonatal (SCREEN) Ileomeconial (MI) História Familiar (FH) Diagnóstico pela clínica (SYMPTOM)
 pacientes com FC divididos em 4 grupos: Diagnóstico prenatal ou neonatal (SCREEN) Ileomeconial (MI) História Familiar (FH) Diagnóstico pela clínica (SYMPTOM)")
55
Acompanhamento de 2 grupos de FC
McKay KO. A influencia da triagem neonatal nos desfechos e evolução da doença pulmonar da FC na Australia. J Pediatr 2005; 147: S47 - S50 Acompanhamento de 2 grupos de FC Com e sem Screning neonatal A função pulmonar e os escores clínicos foram melhores até a adolescência no grupo que teve o diagnóstico por screning neonatal

56
Dankert-Roelse JE et al
Dankert-Roelse JE et al. Resultados da sobrevida de pacientes com FC com e sem a triagem neonatal na Europa. J Pediatr 2005;147:S15-S20 S = Screening

57
Farrell PM et al. Evidence on improved outcomes with early diagnosis of cystic fibrosis through neonatal screening: Enough is enough! J. Pediatr 2005; 147:S30-S36 . Estados Unidos Wisconsin Cystic Fibrosis Neonatal Screening Project

58
Wisconsin Cystic Fibrosis Neonatal Screening Project
Journal of Pediatrics : S30 - S36.

59
A 6ª Vara Federal Cível de São Paulo obrigou a União, o Estado e o Município a implantar e realizar, no prazo de 90 dias, a triagem neonatal para o diagnóstico da “Fibrose Cística” em todos os recém-nascidos vivos no Estado de São Paulo. A notícia completa encontra-se no Blog Mauro Ferreira Gerente Medico-Marketing de Fibrose Cística United Medical Ltda. Visite o Blog

60
Roteiro diagnóstico na fibrose cística
Sintomas clínicos ou história familiar positiva ou triagem neonatal positiva Teste do suor Cloro 60 mmol/L Cloro mmol/L Cloro 40 mmol/L Repetir dosagem Repetir dosagem Investigação adicional somente se sintomas típicos Cloro 60 mmol/L Cloro mmol/L Diagnóstico confirmado Genotipagem para as mutações mais freqüentes Lancet

61
Roteiro diagnóstico na fibrose cística
Genotipagem para as mutações mais freqüentes 2 mutações CFTR 1 mutação CFTR Sem mutação CFTR Repetir exame Testes clínicos adicionais (enzimas pancreáticas fecais, Rx de seios da face, swab de orofaringe ou de escarro, espermograma) Investigação adicional somente se sintomas típicos Diagnóstico confirmado Medida da diferença de potencial nasal ou biópsia de mucosa retal Lancet 2003
 Investigação adicional somente se sintomas típicos. Diagnóstico confirmado. Medida da diferença de potencial nasal ou biópsia de mucosa retal. Lancet")
62
Se alterações da CFTR não puderem ser demonstradas por algum método:
Teste do suor Análise das mutações Medida da diferença de potencial nasal Função CFTR em células do intestino O diagnóstico definitivo não pode ser realizado. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998

63
FC clássica: diagnóstico clínico na maioria dos pacientes apoiada por testes bioquímicos e genéticos O teste do suor continua diagnóstico na maioria dos pacientes Existe um número pequeno, mas crescente, de pacientes que não satisfazem os critérios de diagnóstico padrão desafio diagnóstico! O que é FC não-clássica ou atípica? Diagnóstico difícil

65
O que é FC atípica? Pacientes com uma ou apenas algumas, geralmente leves, características clínicas da fibrose cística Manifestação clínicas na vida adulta ou detecção pela triagem neonatal Teste do suor negativo ou borderline Duas mutações sem sintomas ou uma mutações CFTR com sintomas Vários polimorfismos

66
4) Quadro clínico Fibrose Cística
Rosestein BJ et al. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998

67
Sintomas presentes ao diagnóstico. Cystic Fibrosis Foundation: 20
Sintomas presentes ao diagnóstico. Cystic Fibrosis Foundation: pacientes cadastrados em 1997 nos USA A. Sint. respiratórios: ,5% B. DPC ,9% C. Esteatorréia ,0% D. Ileo meconial ,8% E. História familiar ,8% F. Distúrbio eletrolítico 5 ,4% G. Prolapso retal ,4% H. Screening neonatal ,3% I. Pólipos nasais ,0% J. Genótipo ,2% K. Doença hepatobiliar 0,9% L. Diag. Prénatal ,8%

68
Typical and Atypical Phenotypic Features of CF
Chronic sinusitis Chronic sinusitis Severe chronic infection Chronic infection Severe hepatobiliary disease Normal hepatobiliary function Pancreatic insufficiency Pancreatic sufficiency Meconium ileus at birth No meconium ileus at birth F. Hoffmann-La Roche Ltd does not encourage, condone, or promote alterations, modifications or deletions of this promotional item. Any alteration, modification, or deletion and subsequent consequences would be the sole responsibility of the end-user. Typical and Atypical Phenotypic Features of CF Typically, affected patients exhibit chronic sinopulmonary disease, involving persistent colonization or infection with Staphylococcus aureus, Haemophilus influenzae, Pseudomonas aeruginosa, and/or Burkholderia cepacia. There may also be evidence of nasal polyps or digital clubbing.14 Gastrointestinal abnormalities may be present, such as chronic hepatobiliary disease manifested by biliary or multilobular cirrhosis, pancreatic insufficiency, or meconium ileus.14 Salt-loss syndromes, which may produce elevated sweat chloride values.14,15 Most males with CF have obstructive azoospermia (lack of spermatozoa in the semen as a result of congenital bilateral absence of the vas deferens).14,15 In atypical CF, disease symptoms are less severe and fewer organ systems are affected. In particular, atypical patients are pancreatic sufficient and may have normal or slightly elevated borderline sweat chloride values.15 14. Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998;132: 15. Knowles MR, Durie PR. What is cystic fibrosis? N Engl J Med 2002;347: Elevated sweat chloride values Normal or borderline sweat chloride values Obstructive azoospermia Obstructive azoospermia
.14,15. In atypical CF, disease symptoms are less severe and fewer organ systems are affected. In particular, atypical patients are pancreatic sufficient and may have normal or slightly elevated borderline sweat chloride values Rosenstein BJ, Cutting GR. The diagnosis of cystic fibrosis: a consensus statement. J Pediatr 1998;132: Knowles MR, Durie PR. What is cystic fibrosis N Engl J Med 2002;347: Elevated sweat chloride values. Normal or borderline. sweat chloride values. Obstructive azoospermia. Obstructive azoospermia.")
69
Vários fenótipos na Fibrose Cística:
Hipertripsinemia neonatal, teste do suor normal FC Clássica com insuficiência pancreática e doença sinuso-pulmonar Doença sinuso-pulmonar, suficiência pancreática e teste do suor positivo Doença sinuso-pulmonar, fertilidade masculina, teste do suor normal Apenas Infertilidade masculina Sinusite grave e ausência congênita bilateral dos vasos deferentes (CBAVD) Apenas Pancreatite crônica Aspergilose broncopulmonar alérgica Apenas Teste do suor positivo Nenhum quadro clínico, incluindo cloro no suor normal, mas duas mutações CFTR
 Apenas Pancreatite crônica. Aspergilose broncopulmonar alérgica. Apenas Teste do suor positivo. Nenhum quadro clínico, incluindo cloro no suor normal, mas duas mutações CFTR.")
Apresentações semelhantes













, Eliana Matos (BA),>")



>")


