Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Anomalias Cromossômicas

2
CITOGENÉTICA HUMANA CONCEITOS Para um melhor entendimento do assunto aqui abordado, faz-se necessário um conhecimento prévio dos termos aqui utilizados.

3
conjunto de cromossomos característicos de uma espécie.
Cariótipo: conjunto de cromossomos característicos de uma espécie.

4
longa molécula de DNA associada com proteínas (histona).
Cromossomos: longa molécula de DNA associada com proteínas (histona).
.")
5
Cromossomos Autossomos:
são os cromossomos que equivalem nos indivíduos masculinos e femininos. (22 pares)
")
6
Cromossomos Sexuais ou Heterossomos:
são os cromossomos que se diferem nos indivíduos masculinos e femininos (1 par X e Y).
.")
7
Aberração Cromossômica:
qualquer modificação no cariótipo, podem ser numéricas ou estruturais.

8
CARIÓTIPO HUMANO NORMAL
Tjio e Levan estabeleceram, em 1956, que o número diplóide correto de cromossomos do cariótipo humano era 46, sendo composto de 23 pares de cromossomos homólogos: 22 pares autossomos + XY (no sexo masculino) ou XX (no sexo feminino).
 ou XX (no sexo feminino).")
9
Três anos mais tarde, foi descoberta uma alteração no cariótipo normal, era um trissomia em indivíduos portadores da síndrome de Down (comentada a seguir), descoberta por Lejeune. Foi a partir de então que foi possível explicar aproximadamente 12 síndromes congênitas humanas e demonstrar que cerca de 5 entre 1000 recém-nascidos apresentam algum tipo de aberração cromossômica.

10
TÉCNICAS DE CARIOTIPAGEM
Diversas técnicas são usadas no estudo do cariótipo humano, são empregadas culturas de fibroblastos de medula óssea, sangue periférico e pele. As mitoses são bloqueadas com colchicina (substância que bloqueia a formação dos microtúbulos do fuso) durante a metáfase.
 durante a metáfase.")
11
O cariótipo é habitualmente obtido através de fotomicrografia
O cariótipo é habitualmente obtido através de fotomicrografia. Cada cromossomo é recortado e alinhado com o seu homólogo em uma ordem decrescente de tamanho. Esta técnica é facilitada pela determinação do índice centromérico, razão entre os comprimentos dos braços longos e curtos do cromossomo.

12
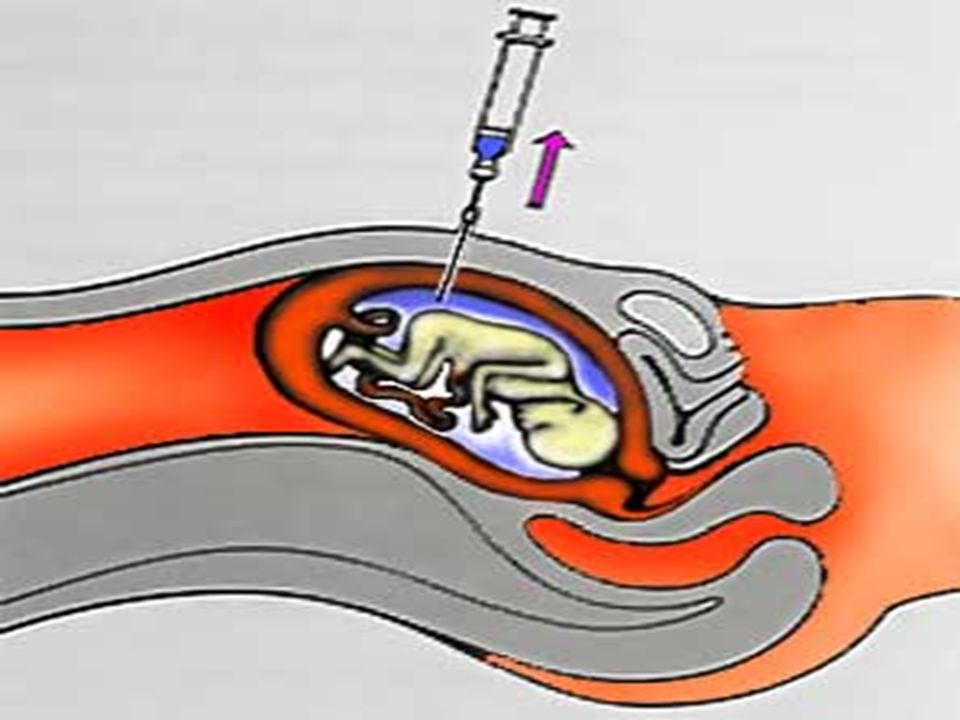
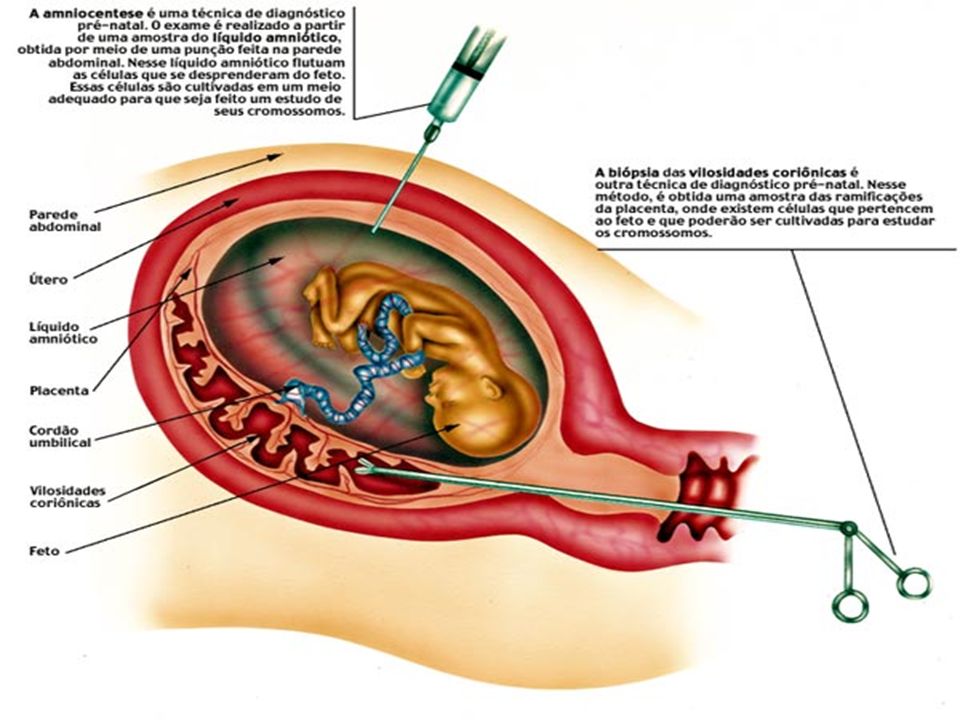
É possível a obtenção de células para cariotipagem e diagnóstico de aberrações cromossômicas mesmo antes do nascimento da criança. Isto é possível através da amniocentese, ou seja, punção da parede uterina com uma agulha para obtenção do fluido amniótico, que é analisado para determinar se o feto possui alguma aberração e até mesmo o sexo deste.

13
CROMATINA X Em 1949, Barr e Bertram descobriram que no núcleo interfásico de células de indivíduos do sexo feminino existe um pequeno corpúsculo de cromatina que não aparece nas células do sexo masculino. Este corpúsculo foi chamado de cromatina sexual ou corpúsculo de Barr e a partir da confer6encia de Paris, cromatina X.

14
A cromatina X pode ser encontrada em diferentes posições no núcleo, ela deriva de um dos cromossomos X presentes na célula, o número de cromatina X é dado por: nX - 1, ou seja, número de cromossomos X menos uma unidade. Por exemplo, um indivíduo normal do sexo feminino (XX) possui apenas 1 cromatina X. nX = 2.
 possui apenas 1 cromatina X. nX = 2. .")
15
ANOMALIAS CROMOSSÔMICAS
O funcionamento normal do sistema genético depende da estabilidade do material genético contido nos cromossomos. O cariótipo pode modificar-se levando a alterações genéticas, tais alterações podem ser no número de cromossomos quanto na sua estrutura. As alterações no número de cromossomos pode levar a euploidia ou aneuploidia.

16
EUPLOIDIAS Você já aprendeu anteriormente que as células somáticas apresentam dois genomas, sendo, portanto, diplóides (2n). Quando ocorre um aumento ou uma diminuição do número de genomas em uma célula, temos uma euploidia. Os casos mais importantes de euploidia são os seguintes: Monoploidia - Quando as células somáticas são haplóides (n), apresentam apenas um genoma.
. Quando ocorre um aumento ou uma diminuição do número de genomas em uma célula, temos uma euploidia. Os casos mais importantes de euploidia são os seguintes: Monoploidia - Quando as células somáticas são haplóides (n), apresentam apenas um genoma.")
17
O exemplo característico é o do zangão, que se origina de partenogênese do óvulo posto por uma rainha, o qual germinará sem ter sido fecundado. Triploidia - A triploidia caracteriza-se pela presença de três genomas (3n) nas células somáticas de um indivíduo. A triploidia é pouco freqüente, mas ocorre, por exemplo, com as células do endosperma das sementes, representando uma reserva nutritiva para o embrião do vegetal. Poliploidia - Ocorre quando as células apresentam quatro ou mais genomas e normalmente resulta da interferência humana, como no caso de plantas cultivadas, como o café (4n), o trigo (6n) e outras.
 nas células somáticas de um indivíduo. A triploidia é pouco freqüente, mas ocorre, por exemplo, com as células do endosperma das sementes, representando uma reserva nutritiva para o embrião do vegetal. Poliploidia - Ocorre quando as células apresentam quatro ou mais genomas e normalmente resulta da interferência humana, como no caso de plantas cultivadas, como o café (4n), o trigo (6n) e outras. .")
18
ANEUPLOIDIAS Neste tipo de mutação, ocorre um aumento ou uma diminuição de um ou mais cromossomos no genoma, alterando o cariótipo daqueles que a apresentam. Alguns dos importantes casos de aneuploidias são: Nulissomia (2n - 2) - Quando está ausente um dos pares de cromossomos homólogos. Devido ao grande número de disfunções e malformações surgidas pela ausência de um par de cromossomos, este tipo de mutação é freqüentemente letal.
 - Quando está ausente um dos pares de cromossomos homólogos. Devido ao grande número de disfunções e malformações surgidas pela ausência de um par de cromossomos, este tipo de mutação é freqüentemente letal.")
19
Monossomia (2n - 1) - Quando falta um cromossomo de um dos pares do conjunto diplóide, ficando o indivíduo com 45 cromossomos. A síndrome de Turner é um caso clássico na espécie humana e se caracteriza pela ausência de um dos cromossomos do par sexual. Esta síndrome apresenta uma freqüência de 1/5000 recém-nascidos do sexo feminino com um cariótipo 44A + X0. Trissomia (2n + 1) - Neste tipo de mutação ocorre a presença de um cromossomo a mais de um dos pares do lote diplóide, ficando os indivíduos com 47 cromossomos.
 - Neste tipo de mutação ocorre a presença de um cromossomo a mais de um dos pares do lote diplóide, ficando os indivíduos com 47 cromossomos..")
20
Um caso bastante conhecido na espécie humana é a síndrome de Down ou mongolismo, que apresenta uma freqüência de 1/800 recém-nascidos independentemente do sexo. Ela caracteriza-se pela presença de um cromossomo a mais no par 21, ficando os indivíduos com os cariótipos 45A + XY = 47 ou 45A + XX = 47. Outra trissomia autossômica conhecida é a síndrome de Edwards, com um cromossomo a mais no par 18. Ela apresenta uma freqüência de 1/6000 nascimentos.

21
Mais uma trissomia bastante conhecida é a síndrome de Klinefelter, com uma freqüência de 1/900 recém-nascidos do sexo masculino, os quais apresentam um cromossomo X a mais no par sexual, ficando com o cariótipo 44A + XXY. As principais causas conhecidas para o surgimento destas e de outras síndromes humanas são, principalmente, a gestação com idade materna avançada, a predisposição genética familiar, além da ação de radiações, drogas e vírus.

22
ALTERAÇÕES NA ESTRUTURA DOS CROMOSSOMOS
A aberração cromossômica é uma desorganização na estrutura cromossômica que pode ser observada ao microscópio. Quando ocorre uma mutação gênica, ocorre alteração na seqüência de bases do DNA; tais alterações podem ser observadas através da expressão do gene, e podem ser intra ou intercromossômicas. As principais aberrações cromossômicas são:

23
Deleção ou deficiência: envolve a perda de material cromossômico;
Duplicação: um segmento do cromossomo é representado duas ou mais vezes, se o fragmento duplicado incluir o centrômero ele pode ser incorporado ao cariótipo como um cromossomo extra; Inversão: envolve a inversão de 180° de um segmento cromossômico

24
PREVENÇÃO AS ANOMALIAS GENÉTICAS

25
Diagnóstico pré-natal

26
Amniocentese Conceito:

27
Amniocentese é um exame realizado para checar a saúde do bebê durante a gravidez. O bebê cresce em seu útero, em uma bolsa de água especial chamada de saco anmiótico. O fluido neste saco é testado para checar diferentes tipos de problemas.

28
O que acontece durante o exame
O que acontece durante o exame? O médico limpará seu abdômen e provavelmente anestesiará sua pele. Uma agulha longa é colocada através de seu abdômen e dentro do útero causando algum desconforto. O médico usará ultra-som para auxiliar a guiar a agulha para longe do bebê e onde a maioria do fluido está retirando-o para mandá-lo ao laboratório.

29
O que acontece posteriormente
O que acontece posteriormente? Você e o bebê serão examinados durante um tempo. Então poderá ir para casa e descansar por 24 horas

30
Quais são as desvantagens de realização do exame
Quais são as desvantagens de realização do exame? Apenas 1 em 100 mulheres têm algum problema. Porém é importante conhecer o que pode acontecer. Amniocentese pode causar: - Muita perda de sangue. - Infecções. - Ferir o bebê, a placenta ou o cordão com a agulha. - Rompimento precoce da bolsa de águas. - Dores de parto ou contrações precoces. - Aborto. (Menos que 1 em 200 mulheres têm aborto devido à amniocentese.)
")
31
Quais as vantagens do exame
Quais as vantagens do exame? Poderá avaliar: - Problemas genéticos do bebê, como a Síndrome de Down. - Defeitos do nascimento como uma espinha bífida. - Infecção do líquido amniótico. - Como os pulmões do bebê irão trabalhar.

32
O que pode ser observado após o teste
O que pode ser observado após o teste? Procure ajuda médica se: - Começar a ter contrações ou cólicas intensas. (É normal ter alguma cólica por um curto tempo). - Estiver sangrando ou com qualquer saída de líquido pela vagina que flui sem parar. (É normal ter uma pequena mancha ou gotejamento primeiramente). - Tiver febre. - Tiver qualquer mudança que preocupe. - Tiver perguntas sobre o exame ou seus resultados. - Quiser marcar outra consulta.
. - Estiver sangrando ou com qualquer saída de líquido pela vagina que flui sem parar. (É normal ter uma pequena mancha ou gotejamento primeiramente). - Tiver febre. - Tiver qualquer mudança que preocupe. - Tiver perguntas sobre o exame ou seus resultados. - Quiser marcar outra consulta.")
35
Diagnóstico pré-natal

36
SÍNDROMES

37
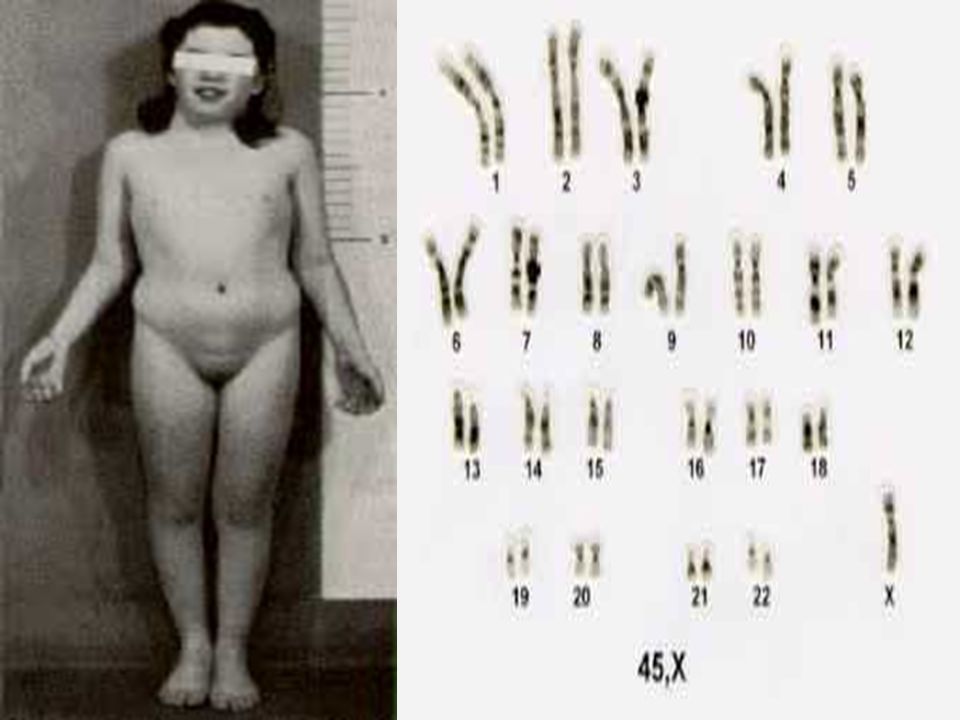
SINDROME DE TURNER

39
A Síndrome de Turner é uma anomalia sexual cromossômica, cujo cariótipo é 45, X, sendo portanto, encontrada em meninas. A Síndrome de Turner, ao contrário de outras aneuploidias dos cromossomos sexuais, é identificada ao nascimento ou antes da puberdade por suas características fenotípicas distintivas. A incidência do fenótipo da Síndrome é de cerca de 1 em 5000 meninas nativivas.

40
A constituição cromossômica mais constante é 45, X sem um segundo cromossomo sexual, X ou Y. Contudo, 50% dos casos possuem outros cariótipos. Um quarto dos casos envolve cariótipos em mosaico, nos quais apenas uma parte das células é 45, X.

41
Fenótipo: BAIXA ESTATURA; DISGENESIA GONADAL; FÁCIES INCOMUM TÍPICA;
DISGENESIA GONADAL; FÁCIES INCOMUM TÍPICA; PESCOÇO ALADO; LINHA POSTERIOR DE IMPLNTAÇÃO DOS CABELOS BAIXA; TÓRAX LARGO COM MAMILOS AMPLAMENTE ESPAÇADOS; FREQÜÊNCIA ELEVADA DE ANOMALIAS RENAIS E CARDIOVASCULARES.

42
Essa anormalidade é responsável 18% dos abortos espontâneos cromossomicamente anormais e está presente numa proporção estimada em 1,5% dos conceptos. O único X é de origem materna; em outras palavras, o erro meiótico costuma ser paterno.

43
Na idade adulta, muitas pacientes com Síndrome de Turner se afligem por sua infertilidade e baixa estatura. Embora a terapia com estrogênios possa levar ao desenvolvimento dos órgãos genitais internos e externos, caracteres sexuais secundários e menstruações, não corrige a infertilidade, que é uma característica quase constante, reultado da atresia das células germinativas iniciais.

44
Atualmente, se estuda o possível valor de baixas doses de estrogênio, androgênio e hormônio do crescimento na terapia da baixa estatura na Síndrome de Turner. Até agora, poucos estudos envolvendo grandes números de pacientes forneceram dados sobre o impacto desses agentes na estatura adulta final, mas está claro que cada droga pode afetar a taxa de crescimento a curto prazo.

45
SINDROME DE KLINEFELTER

47
São indivíduos do sexo masculino que apresentam cromatina sexual e cariótipo geralmente 47,XXY. Eles constituem um dentre 700 a 800 recém-nascidos do sexo masculino, tratando-se, portanto; de uma das condições intersexuais mais comuns. Outros cariótipos menos comuns são 48 XXYY; 48,XXXY; 49,XXXYY e 49,XXXXY que, respectivamente, exibem 1, 2. e 3 corpúsculos de Barr. Embora possam ter ereção e ejaculação. são estéreis, pois seus testículos são pequenos e não produzem espermatozóides devido à atrofia dos canais seminíferos. Outras características multas vezes presentes são: estatura elevada corpo eunucóide, pênis pequeno, pouca pilosidade no púbis e ginecomastia (crescimento das mamas).
..")
48
Além dessas alterações do sexo fenotípico os pacientes com Síndrome de Klinefelter apresentam uma evidente diminuição do nível Intelectual, sendo esta tanto mais profunda quanto maior for o grau da polissomia. Ao contrario do que ocorre na Síndrome de Turner, os pacientes Klinefelter apresentam problemas no desenvolvimento da personalidade, que é Imatura e dependente, provavelmente em decorrência de sua inteligência verbal diminuída. As dificuldades de relacionamento interpessoal incluem, por vezes, alterações no processo de Identificação psicossexual, envolvendo casos de transvestismo, homossexualismo e transexualismo. Fisicamente são quase indistinguíveis dos homens com cariótipo 46,XX, que foram mencionados no capítulo 8.

49
Até 1960 a prova definitiva para o diagnóstico era fornecida pelo exame histológico dos testículos que, mesmo após a puberdade, revela ausência de células germinativas nos canais seminíferos; raros são os casos de Klinefelter férteis que, evidentemente, apresentam alguns espermatozóides normais. Atualmente a Identificação dos Klinefelter é assegurada pelo cariótipo e pela pesquisa da cromatina sexual.

50
Os pacientes são altos e magros, com membros inferiores relativamente longos. Após a puberdade os sinais de hipogonadismo se tornam óbvios. Os testículos permanecem pequenos e os caracteres sexuais secundários continuam subdesenvolvidos.

51
Jovem do sexo masculino, 17 anos, com síndrome de Klinefelter
Jovem do sexo masculino, 17 anos, com síndrome de Klinefelter. Principal Queixa, ginecomastia

52
Paciente B. H. M., com síndrome de Klinefelter

53
SÍNDROME DE EDWARDS

56
Síndrome de Edwards Primeiramente descrita em 1960 por Edwards e colaboradores, a trissomia do 18 é caracterizada também por malformações múltiplas como: maxilar inferior retraído, dedos flexionados ou cerrados, defeitos cardíacos, deformidades do crânio, da face e dos pés. Registram-se, ainda, lábio leporino e palato fendido. As crianças com triplo 18 apresentam grave retardo mental e a morte ocorre geralmente em cerca de 3 a 4 meses de idade.

57
A maioria dos pacientes apresentam com a trissomia do cromossomo 18 apresenta trissomia regular sem mosaicismo, isto é , cariótipo 47, XX ou XY, +18. Entre os restantes, cerca de metade é constituída por casos de mosaicismo e outro tanto por situações mais complexas, como aneuploidias duplas, translocações.

58
Região occipital alongada

59
Sobreposição de artelhos
Sobreposição do segundo e quinto dedo sobre o terceiro e quarto (Figura 2) , assim como pé de balanço foram encontrados em todos os pacientes avaliados Sobreposição de artelhos
 , assim como pé de balanço foram encontrados em todos os pacientes avaliados. Sobreposição de artelhos.")
60
Micrognatia Microretrognatia, implantação baixa das orelhas e occipício proeminente

61
Micrognatia

62
SINDROME DE DOWN

64
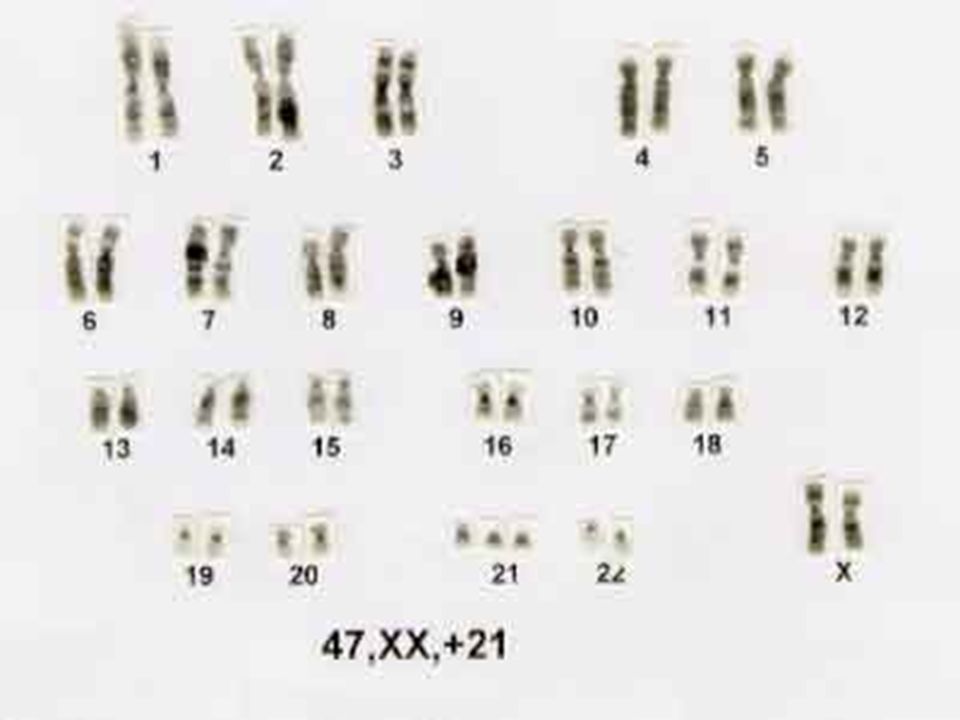
A Sindrome de Down ou trissomia do 21, é sem dúvida o distúrbio cromossômico mais comum e a mais comum forma de deficiência mental congênita. Geralmente pode ser diagnosticada ao nascimento ou logo depois por suas características dismórficas, que variam entre os pacientes, mas produzem um fenótipo distintivo.

65
Devida à trissomia do cromossomo 21 e ocorre com a freqüência de 1 caso para 500 a 600 nascimentos normais. Causa, seguramente, mais deficientes mentais do que qualquer outra doença. A síndrome de Down constituiu a primeira anormalidade cromossômica descrita para o homem, pois Lejeune, já em 1959, demonstrou que os indivíduos com síndrome de Down apresentam excesso de um pequeno cromossomo acrocêntrico. De acordo com a classificação de Denver, trata-se de um cromossomo do grupo G; porém, é impossível saber com certeza se se trata do cromossomo 21 ou do 22, pois ambos são morfologicamente Idênticos. Em todo caso, deve tratar-se sempre do mesmo cromossomo que se encontra triplicado em todos os mongolóides; por convenção, este cromossomo é considerado como sendo o 21.

66
A identificação dos mongolóides, principalmente dos brancos, é fácil pois a aparência dos pacientes é típica. A 'característica mais marcante é o retardamento mental, pois seu QI varia entre 15 e 50. Outras características são a face achatada, a existência de uma prega típica no canto dos olhos, a língua saliente e sulcada, a dentição irregular, as orelhas pequenas e deformadas. O abdômen costuma ser saliente e o tecido adiposo é abundante. A genitália é pouco desenvolvida; nos homens o pênis é pequeno e há criptorquidismo e nas mulheres os lábios e o clitóris são pouco desenvolvidos; embora não se conheça nenhum caso de homem afetado que tenha se reproduzido, as mulheres mongolóides são férteis.

67
Os dedos são, freqüentemente, curtos e grossos com falta de uma falange no dedo mínimo; nas palmas das mãos é comum a existência de uma prega transversal denominada prega simiesca. A pele é flácida determinando o aparecimento de rugas nas frontes e os ligamentos são frouxos causando uma marcha insegura; defeitos do coração são freqüentes. Em conseqüência das anomalias cardíacas e de uma baixa resistência a infecções, a longevidade dos mongolóides costumava ser reduzida; hoje os cuidados médicos aumentam sensivelmente as probabilidades de sobrevivência dos mongolóides.

68
No recém-nascido não se nota o principal sintoma de mongolismo, ou seja, o retardamento mental. Peso inferior ao normal, orelha disforme, prega simiesca e outros achados na mão dos recém-nascidos são algumas indicações que recomendam a realização do exame citogenético.

69
Os pés mostram um amplo espaço entre o primeiro e o segundo dedos.
As mãos são curtas e largas, frequentemente com uma única prega palmar transversa ("prega simiesca"). Os pés mostram um amplo espaço entre o primeiro e o segundo dedos.
. Os pés mostram um amplo espaço entre o primeiro e o segundo dedos.")
70
Aparencia facial de uma paciente com SD.
Portadores de SD: raça negra e amarela. Perfil achatado

71
Olhos com fendas palpebrais oblíquas
Língua grande, protrusa e sulcada Orelhas pequenas

72
Encurvamento dos quintos dígitos
Aumento da distância entre o primeiro e o segundo artelho Prega única nas palmas.

73
Risco aproximado de nascimento da criança com Síndrome de Down no caso de mães de diversas idades, que nunca tiveram uma criança com esta Síndrome Risco aproximado de nascimento da criança com Síndrome de Down no caso de mães de diversas idades, que já tiveram uma criança com esta Síndrome Idade da mãe ao nascer a criança: Risco de nascer criança com Síndrome de Down menos de 35 anos 0,1% 1,0% de 35 a 39 anos 0,5% 1,5% de 40 a 44 anos 2,5% acima de 45 anos 3,5% 4,5%

74
SÍNDROME DE PATAU

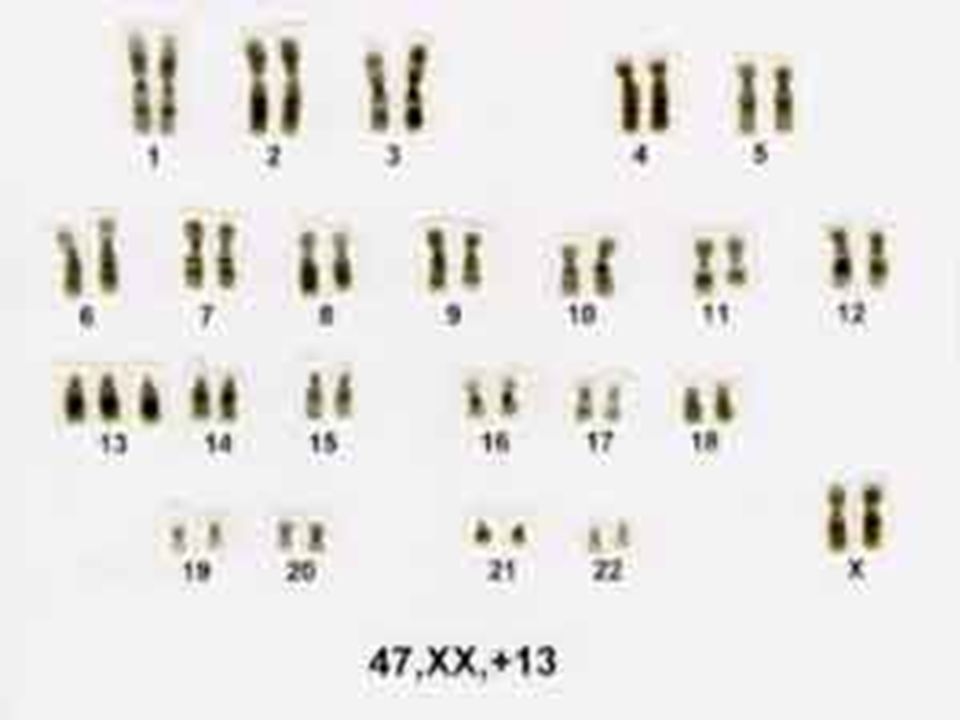
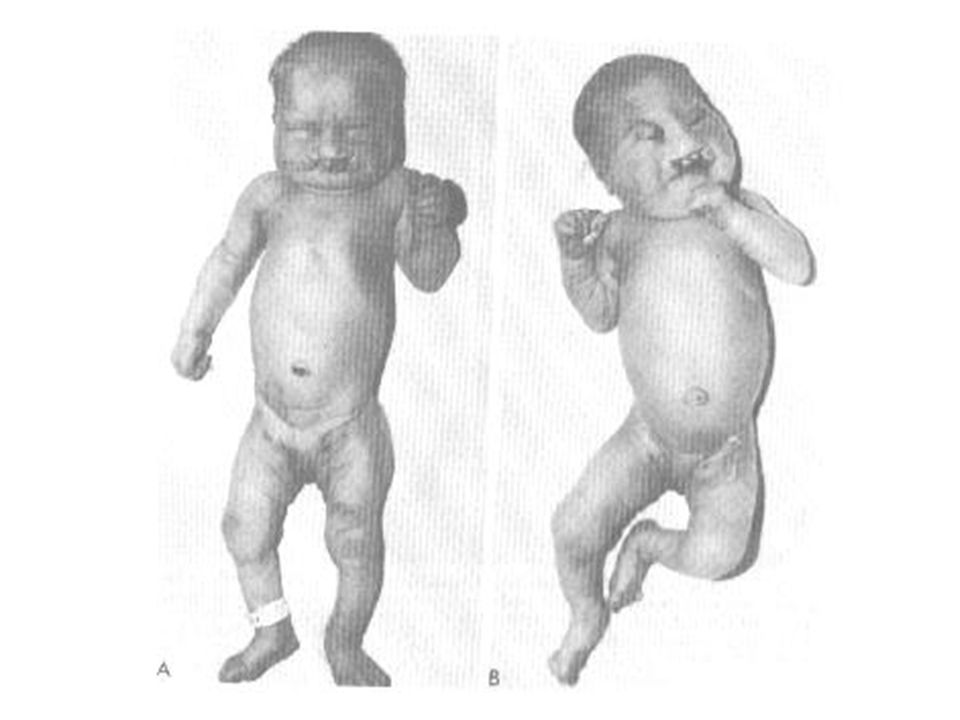
76
A trissomia do 13 é clinicamente grave e letal em quase todos os casos que sobrevivem até 6 meses de idade. O cromossomo extra provém de não-disjunção da meiose I materna e cerca de 20% dos casos resultam de uma translocação não-balanceada.

77
O fenótipo inclui malformações graves do sistema nervoso central como arrinencefalia. Um retardamento mental acentuado está presente. Em geral há defeitos cardíacos congênitos e defeitos urigenitais. Com frequência encontram-se fendas labial e palatina, anormalidades oculares, polidactilia, punhos cerrados e as plantas arqueadas.

78
1 . Trissomia do 13: A trissomia do cromossomo 13 tem como definição uma desordem cromossomal resultando em uma síndrome caracterizada especificamente pôr anomalias morfológicas e malformação de órgãos, gerando a inviabilidade dos afetados. Ocorre quando existem três cromossomos 13 em lugar do par normal no genótipo de um recém nascido. Tem como causa a não disjunção dos cromossomos durante a anáfase 1, gerando gametas com 24 cromátides.

79
2. Histórico: Observada na literatura a primeira vez em 1657 pôr Bartholin, e descrita em 1960 pôr Patau e colaboradores, que a denominaram trissomia do cromossomo D1. Logo em seguida, a síndrome determinada pôr essa aneuploidia foi minuciosamente estudada pôr diversos autores, de sorte que, e, em pouco tempo, ela pôde ser caracterizada clinicamente com bastante precisão. Estudos auto-radiográficos e de fluorescência forneceram evidências de que o cromossomo trissômico nesta síndrome é o 13.

80
4. Patogênese Genética: Quadro clínico rico em sinais e cerca de 75% dos casos apresentam cariótipo com trissomia regular. A trissomia é devida ao fato de não haver disjunção dos cromossomos durante a anáfase 1 da meiose A gravidez em idade avançada aumenta a possibilidade da não disjunção dos cromossomos. Sua taxa de ocorrência é 1/4.000.

81
5. Diagnóstico: O diagnóstico clínico da síndrome de Patau, a ser confirmado pelo exame cromossômico é fácil, pois dentre os seus sinais mais comuns estão: Baixo peso corporal (2.600Kg); Microcefalia e testa achatada; Hipertelorismo ocular e microftalmia bilateral, podendo chegar a anoftalmia; Lábio leporino acompanhado ou não de palatosquise ou palato alto; Queixo pequeno; Defeitos na face média e cérebro anterior; Orelhas dismórficas de implantação baixa e surdez aparente;
; Microcefalia e testa achatada; Hipertelorismo ocular e microftalmia bilateral, podendo chegar a anoftalmia; Lábio leporino acompanhado ou não de palatosquise ou palato alto; Queixo pequeno; Defeitos na face média e cérebro anterior; Orelhas dismórficas de implantação baixa e surdez aparente;")
82
Pescoço curto; Fronte inclinada; Hemangiomas planos na cabeça; Distância intermamilar grande; Apnéias prolongadas; Cardiopatias congênitas, que representam comunicação interventricular e persistência do conduto arterial; Apêndice pré-sacral e fóvea coccigeana; Hérnia inguinal ou umbilical; Genitais externos anômalos (criptorquidia escroto, abdominal, genitália ambígua e pênis encurvado entre os meninos, e clitoromegalia e vagina dupla entre as meninas);
;")
83
Mãos com hexadactilia uni- ou bilateral, geralmente com o polegar e os dois últimos dedos sobrepostos aos outros; unhas estreitas e hiperconvexas; Prega de flexão palmar única, trirrádio axial em posição bastante distal (t’’ e t ’’’) e arco na região tenar; Pés com hexadactilia uni- ou bilateral e com região plantar convexa (pés em cadeira de balanço); Arco ou arco retorcido em S na região halucal. Arrinencefalia (ausência de bulbo e trato olfativo);
 e arco na região tenar; Pés com hexadactilia uni- ou bilateral e com região plantar convexa (pés em cadeira de balanço); Arco ou arco retorcido em S na região halucal. Arrinencefalia (ausência de bulbo e trato olfativo);")
84
Deficiência mental; Útero bicorne; Rim policístico, hidronefrose, hidroureter e ureteres duplos, relacionados a oligúria e anúria em afetados; Atrofia ou ausência das últimas costelas e de vértebras, e hiperplasia sacral; Presença de Hemoglobina Gower 2, que é uma hemoglobina embrionária que desaparece no terceiro mês de gestação; Neutrófilos com núcleo mostrando muitas saliências pedunculadas ou sésseis.

85
A trissomia tem origem do óvulo feminino, pelo fato da fêmea maturar geralmente apenas um ovócito, em antagonismo com o macho, que matura milhões de espermatozóides. Gametas masculinos portadores de alterações numéricas cromossômicas tem menor viabilidade que gametas normais, sendo mínimas as possibilidades de um gameta masculino com 24 cromátides fecundar um ovócito.

88
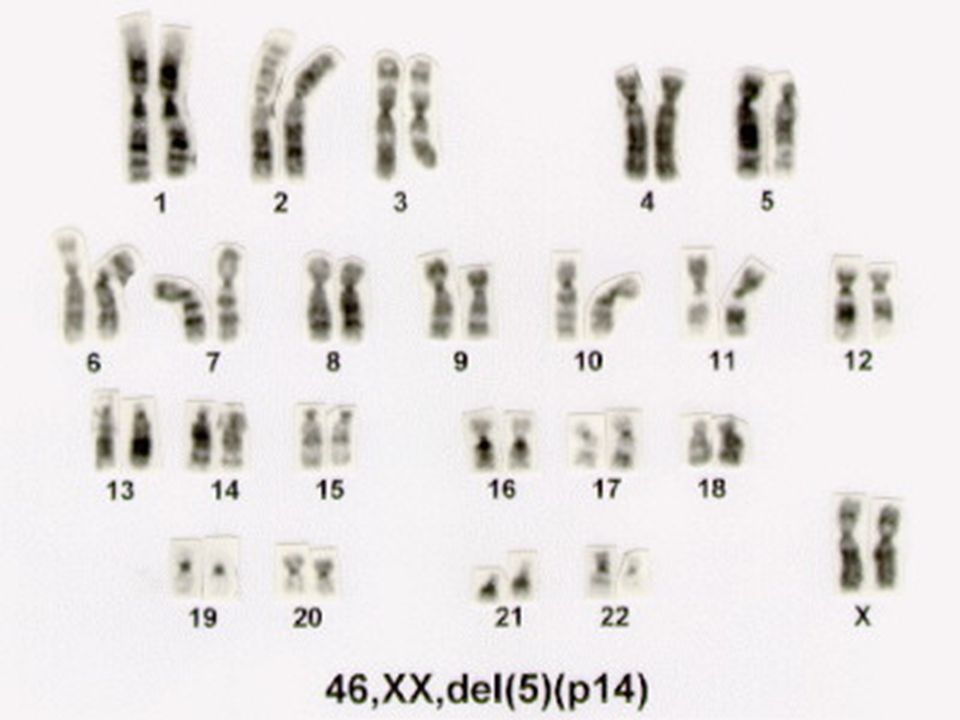
SÍNDROME MIADO DE GATO

90
46, XX ou XY, 5p- (deleção no braço curto do cromossomo 5)
Peso Baixo ao nascer, crescimento lento Deficiência mental Hipotonia Microcefalia

91
Face Arredondada Hipertelorismo Choro como o miado do gato. 85% resultam de uma nova deleção. freqüência 1: nascimentos

92
INTRODUÇÃO Em 1963, o Dr. Lejeune na França descobriu a Síndrome do Miado de Gato. Foi dado este nome devido ao choro característico lamentoso, muito similar ao de um pequeno gato em sofrimento.Este sintoma está associado a uma malformação da laringe. Outras malformações características envolvem a cabeça e face. A síndrome do miado de gato é uma anomalia cromossômica, causada pela deleção (quebra) da perna curta do cromossomo 5. Por isso é também chamada de síndrome 5 p - (menos). Os estudos citológicos indicam a associação dos sintomas principais com a deleção de pequena parte da banda 5p14p15.
 da perna curta do cromossomo 5. Por isso é também chamada de síndrome 5 p - (menos). Os estudos citológicos indicam a associação dos sintomas principais com a deleção de pequena parte da banda 5p14p15.")
93
A estimativa calculada pelos bancos de dados internacionais é de que 1 em crianças nascem com síndrome do miado de gato no mundo. Esta síndrome na maioria das vezes, não é herdada dos pais, somente em 20% dos casos. Esses casos são causados pela translocação equilibrada nos cromossomos de um dos pais (material genético de um cromossomo que se uniu a outro). As pessoas com translocações equilibradas são perfeitamente normais porque nenhum material genético foi perdido, assim sendo, provavelmente não saberão que são portadores até que tenham uma criança afetada com a síndrome na família
. As pessoas com translocações equilibradas são perfeitamente normais porque nenhum material genético foi perdido, assim sendo, provavelmente não saberão que são portadores até que tenham uma criança afetada com a síndrome na família.")
94
CARACTERÍSTICAS Bebês Choro característico logo ao nascer parecendo um miado de gato. Peso baixo ao nascimento, apesar de estarem no período certo gestacional. Hipotonia (tônus muscular deficiente). Atraso nos desenvolvimentos cognitivo e motor. Microcefalia (cabeça pequena) Cara redonda em forma de lua Alguns nascem com problemas cardíacos e/ou renais. Epicanto. Prega única na palma da mão. Orelhas mais baixas que a linha do nariz. Dedos longos.
. Atraso nos desenvolvimentos cognitivo e motor. Microcefalia (cabeça pequena) Cara redonda em forma de lua. Alguns nascem com problemas cardíacos e/ou renais. Epicanto. Prega única na palma da mão. Orelhas mais baixas que a linha do nariz. Dedos longos.")
95
Ao contrário do que a literatura médica antiga dizem sobre as crianças com síndrome do miado de gato, elas não morrem cedo. O tempo de vida espera-se ser relativamente normal, entretanto observa-se alguns sinais de envelhecimento precoce. * É fundamental deixar claro que nem todas as pessoas com síndrome do miado de gato terão todas estas características.

96
DIFICULDADES COGNITIVAS
Crianças com síndrome do miado de gato têm algum grau de déficit cognitivo, variando de moderado a severo, com um quociente de inteligência geralmente inferior a 20. Isso afeta em muito suas vida porque levarão muito mais tempo para aprender. Associado a isto, eles têm tônus muscular fraco, e algumas coisas se tornam muito difíceis. Porém, eles vão aprender ao longo da vida.

97
A aquisição da fala é um grande problema
A aquisição da fala é um grande problema. A fala é atrasada, e para muitas crianças com síndrome do miado de gato dependendo da dificuldade, poderão nunca falar. Entretanto, compreendem o que falam, e se comunicam muito bem, da sua maneira (usando seus próprios signos de linguagem) ou usando sinais, placas, cartazes, fotos etc...
 ou usando sinais, placas, cartazes, fotos etc...")
98
DIFICULDADE COM A ALIMENTAÇÃO
Muitos bebês têm muitas dificuldades de alimentação como pouca força para mamar, ou problemas de refluxo. Algumas crianças mais velhas podem ter dificuldade em mastigar, necessitando que todo o alimento seja preparado com uma consistência adequada.

99
DIFICULDADES MOTORAS Demoram para aprender a andar. As crianças do síndrome do miado de gato freqüentemente têm um caminhar desajeitado e parecem inábeis. As habilidades motoras finas são atrasadas também, embora algumas crianças estejam conseguindo aprender a escrever.

100
OUTRAS DIFICULDADES As crianças com síndrome do miado de gato têm dificuldade no treinamento do controle de suas necessidades fisiológicas. Muitas pessoas com síndrome do miado de gato têm prisão de ventre. A maioria de adultos poderá controlar seu cuidado pessoal com uma supervisão mínima.

101
Muitos bebês e crianças com síndrome do miado de gato têm um sono agitado, mas isto melhora com idade. Muitas crianças com síndrome do miado de gato podem ter problemas de comportamento. Eles podem ser hiperativos, balançam muito a cabeça, podem até dar mordidas ou se beliscarem. Alguns desenvolvem obsessões com determinados objetos. Muitos têm um fascínio por cabelo e não podem resistir a puxá-lo.

102
É muito difícil predizer como cada criança em particular se desenvolverá, pois isso não está apenas relacionado à quantidade de material genético que foi perdida, mas também a estímulos cognitivos, motores e principalmente muito amor de todos os que a cercam.

103
DIAGNÓSTICO Em estudos citológicos, na maioria, senão na totalidade dos casos, a deleção surge durante a gametogênese de um genitor ou do outro. Ambos os genitores da criança afetada apresentam cariótipos normais. Não há um efeito de idade dos genitores.

104
Há um uso crescente de culturas de células do liquido amniótico, obtidas por amniocentese, para se determinar os cariótipos dos fetos. As células amnióticas parecem apresentar uma freqüência incomumente alta de tetraploidia, não acompanhada de nenhuma anomalia citológica no feto, mas as aberrações estruturais são facilmente detectadas nessas células por volta da 16ª semana de gestação. Esta informação pode ser extremamente útil para os genitores e para os médicos, ao se levar em consideração o possível termino da gestação.

105
TRATAMENTO Uma anomalia genética não pode ser curada ainda neste momento. Mas quem sabe com o acelerado desenvolvimento desta área da Ciência (Genoma-estudo de todos os genes). Podemos ter esperança!
. Podemos ter esperança!")
106
Assim sendo depende de todas as pessoas que convivem com ascrianças com síndrome do miado de gato para ajudá-las a se desenvolverem em tudo que for possível. A estimulação precoce é necessária, o tratamento fisioterapêutico, fonoaudiológico e terapêutico ocupacional é imprescindível desde muito cedo. É muito importante que todos lhes dêem muito amor e confiança.

107
Parece que tudo é muito difícil, entretanto as crianças com síndrome do miado de gato, que foram tratadas com amor e carinho são felizes, saudáveis e sociais. Amam a vida e nos ensinam muito. Ensinam a ver a vida de uma outra forma, com muito mais vontade de romper todos os desafios que nos são colocados pela frente

Apresentações semelhantes







 Um processo especial.>")















