Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Fenilcetonúria … … um caso clínico
Faculdade de Medicina da Universidade de Coimbra Bioquímica II SO3 Fenilcetonúria … … um caso clínico Joana Abrantes | Joana Couto | João Barros | João Costa | João Gomes | João Miranda | João Pedro | João Martins

2
índice A doença Como surge? Catabolismo da fenilalanina
Tipos de fenilcetonúria Sintomatologia Diagnóstico Prevenção Tratamento Caso clínico Questões Referências

3
Fenilcetonúria (PKU) É uma doença metabólica, maioritariamente hereditária (autossómica recessiva), originada por uma alteração no gene, presente no cromossoma 12, que codifica o enzima fenilalanina hidroxilase (PAH). É a doença metabólica congénita mais frequente; é também incurável. fenilalanina no sangue bem como de fenilpiruvato (derivado da fenilalanina por transaminação). O nome provém da excreção de ácido fenilpirúvico, uma fenilcetona, na urina. A esperança de vida dos fenilcetunúricos não tratados é muito reduzida! Cerca de ½ dos pacientes está morta aos 20 anos; ¾ dos restantes falecem aos 30 anos. A dita alteração no gene da enzima PAH provoca um défice na produção desta, cuja função é transformar fenilalanina em tirosina. Sem a acção da enzima, a fenilalanina não é transformada em tirosina, ocorrendo antes uma reacção alternativa que conduz à acumulação de certas substâncias nocivas.
, originada por uma alteração no gene, presente no cromossoma 12, que codifica o enzima fenilalanina hidroxilase (PAH). É a doença metabólica congénita mais frequente; é também incurável. fenilalanina no sangue bem como de fenilpiruvato (derivado da fenilalanina por transaminação). O nome provém da excreção de ácido fenilpirúvico, uma fenilcetona, na urina. A esperança de vida dos fenilcetunúricos não tratados é muito reduzida! Cerca de ½ dos pacientes está morta aos 20 anos; ¾ dos restantes falecem aos 30 anos. A dita alteração no gene da enzima PAH provoca um défice na produção desta, cuja função é transformar fenilalanina em tirosina. Sem a acção da enzima, a fenilalanina não é transformada em tirosina, ocorrendo antes uma reacção alternativa que conduz à acumulação de certas substâncias nocivas.")
4
Fenilcetonúria Cerca de 1% dos pacientes internados em instituições mentais têm PKU. O peso do seu cérebro está abaixo do normal; A mielinização dos nervos é deficitária; Reflexos hiperactivos.

5
Como surge? A fenilcetonúria é causada por mutações no gene PAH, que está localizado no cromossoma 12. Esta é uma doença muito heterogénea a nível molecular com mais de 500 mutações diferentes descritas. A maioria das mutações descritas no gene PAH corresponde a substituições nucleotídicas do tipo missense e localiza-se no exão 7, enquanto que as delecções e inserções representam 12% do tipo de mutações observadas.

6
Tyr não essencial, Phe essencial
A fenilalanina (Phe) e a tirosina (Tyr) são ambos AA aromáticos, cetogénicos e glucogénicos; Tyr não essencial, Phe essencial Fenilalanina: Aminoácido aromático, essencial, cetogénico e glucogénico. É normalmente convertido pela enzima fenilalanina hidroxilase no aminoácido tirosina. Posteriormente, a tirosina vai entrar na constituição de novas proteínas ou ser convertida noutras substâncias como fumarato, melanina e catecolaminas (epinefrina, norepinefrina e dopamina).
 e a tirosina (Tyr) são ambos AA aromáticos, cetogénicos e glucogénicos; Tyr não essencial, Phe essencial. Fenilalanina: Aminoácido aromático, essencial, cetogénico e glucogénico. É normalmente convertido pela enzima fenilalanina hidroxilase no aminoácido tirosina. Posteriormente, a tirosina vai entrar na constituição de novas proteínas ou ser convertida noutras substâncias como fumarato, melanina e catecolaminas (epinefrina, norepinefrina e dopamina).")
7
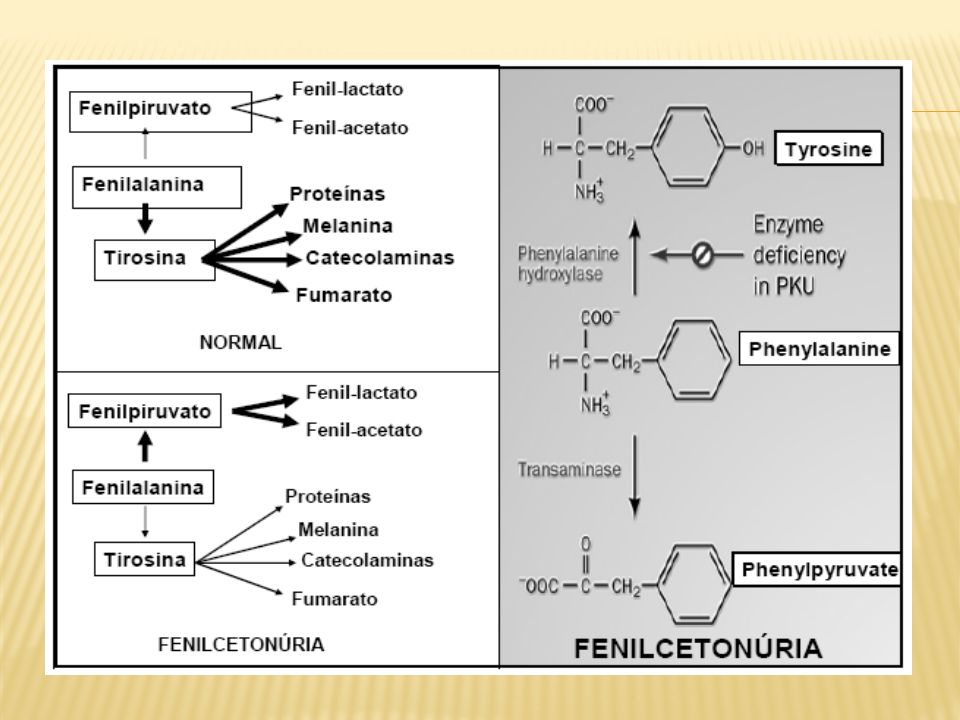
Fenilcetonúria

8
Catabolismo da fenilalanina
Enzima que degrada a fenilalanina em excesso, catalisando a reacção de adição de um grupo hidroxilo ao carbono-6 do anel aromático da fenilalanina, convertendo-a em tirosina; É um tetrâmero, sendo composto por dois dímeros de duas subunidades cada. Cada subunidade requer um átomo de ferro para que a enzima possa exercer actividade catalítica; Tem a 5,6,7,8-tetrahidrobiopterina como co-factor; O défice de actividade desta enzima é a causa da doença fenilcetonúria.

9
Papel da BH4 na reacção da PAH

10
Via catabólica alternativa da Fenilalanina
Responsável pelo odor fétido (“mouse-like odor”) da urina Na ausência da enzima fenilalanina hidroxilase, a fenilalanina não é transformada em tirosina. - É activada uma via alternativa de metabolização da fenilalanina :
 da urina. Na ausência da enzima fenilalanina hidroxilase, a fenilalanina não é transformada em tirosina. - É activada uma via alternativa de metabolização da fenilalanina :")
12
Tipos de PKU Fenilcetonúria tipo I: deficiência na actividade da enzima fenilalanina hidroxilase. PKU clássica: A actividade enzimática é menor que 1%, o que faz com que a concentração de fenilalanina sanguínea fique maior do que 20mg/dl, quando o normal seria de 2 a 6mg/dl. É responsável por quase dois terços dos casos de PKU. PKU leve: Ocorre quando a actividade enzimática é de 1 a 3% e os níveis plasmáticos de fenilalanina encontram-se entre 10 e 20 mg/dl. PKU permanente ou benigna: A actividade enzimática é superior a 3% e os níveis de substrato encontram-se entre 4 e 10mg/dl; situação em que não ocorre qualquer sintomatologia clínica.

13
Fenilcetonúria tipo II: Causada pela deficiência de dihidrobiopterina reductase, a enzima que regenera a tetrahidrobiopterina, essencial para a fenilalanina hidroxilase. Fenilcetonúria tipo III: Causada por um defeito no gene de uma das enzimas envolvidas na biossíntese da tetrahidropioterina. Como a coenzima biopterina é necessária para a hidroxilação da fenilalanina, tirosina e triptofano, a PKU tipo III tem sérios efeitos sobre outras vias metabólicas.

14
Sintomatologia Esta doença apresenta os seguintes sintomas, caso a dieta não esteja a ser posta em prática. Identificam-se as alterações com cerca de 1 ano de vida. Eczema Descamação da pele Níveis baixos de melanina (pela clara, cabelos loiros e olhos azuis) QI inferior a 50 Odor corporal característico resultante da formação de finilalanina no cabelo, pele e urina Oligofrenia Convulsões Atraso de desenvolvimento psicomotor (andar ou falar) Hiperactividade Microcefalia Tremor
 QI inferior a 50. Odor corporal característico resultante da formação de finilalanina no cabelo, pele e urina. Oligofrenia. Convulsões. Atraso de desenvolvimento psicomotor (andar ou falar) Hiperactividade. Microcefalia. Tremor.")
15
Diagnóstico É feito (3 a 6 dias após nascimento) pelo Teste do Pezinho (Prova de Guthrie).
 pelo Teste do Pezinho (Prova de Guthrie).")
16
Diagnóstico - Neonatal
Idealmente o diagnóstico deve ser realizado através do teste do pezinho pois permite um tratamento precoce e prevenção do desenvolvimento do quadro clínico. O teste do pezinho é um exame laboratorial considerado simples que tem como objectivo detectar doenças congénitas, relacionadas com distúrbios do metabolismo e infecções que são realizadas pela analise de gotas de sangue do recém-nascido. As alterações na urina surgem posteriormente às alterações detectadas no sangue, muitas vezes juntamente com os primeiros sinais de lesão do sistema nervoso. Assim, a triagem de fenilcetonúria através da urina é inadequada para um programa de diagnóstico precoce.

17
Diagnóstico Clínico A fenilcetonúria não apresenta sintomas no nascimento. Estes instalam-se nas primeiras semanas ou meses quando não é realizado tratamento. Diferentes mutações génicas determinam diferentes quadros clínicos mas com a maior parte das manifestações constantes. Os sintomas mais frequentes da doença não tratada são: vómitos, eczemas, dermatites, rush cutâneo, odor característico na urina e suor (urina de rato) e despigmentação da pele (com olhos e cabelos mais claros que a família). Entre as manifestações neurológicas destacam-se: irritabilidade, choro frequente, atraso no desenvolvimento neuropsicomotor, convulsões e, principalmente retardo mental. É importante realçar que o tratamento pode e deve ser iniciado antes do aparecimento de qualquer manifestação clínica sendo, portanto, preventivo.
 e despigmentação da pele (com olhos e cabelos mais claros que a família). Entre as manifestações neurológicas destacam-se: irritabilidade, choro frequente, atraso no desenvolvimento neuropsicomotor, convulsões e, principalmente retardo mental. É importante realçar que o tratamento pode e deve ser iniciado antes do aparecimento de qualquer manifestação clínica sendo, portanto, preventivo.")
18
Prevenção Mulheres com fenilcetonúria, devem seguir uma dieta baixa em fenilalanina, especialmente antes e durante a gravidez. Amniocentese (método utilizado no diagnóstico) Monitorização e Controlo Criança Saudável
 Monitorização e Controlo. Criança Saudável.")
19
tratamento Baseia-se numa dieta, que deve ter um controlo rigoroso durante toda a vida. O seguimento deve ser contínuo. A dieta pobre em fenilalanina, por isso, devem evitar-se alimentos como: Carnes vermelhas Ovos Aves Peixe Leite e queijo aspartame

20
Apresentação do Caso Clínico
História Pregressa Criança de 6 meses Desde os 3-4 meses mostra desinteresse pelo mundo que a rodeia apresenta tremor das extremidades apresenta atraso no desenvolvimento psicomotor Recorreu ao hospital por apresentar movimentos de torsão e convulsões

21
Apresentação do Caso Clínico
Antecedentes Pessoais Segundo filho de um casal jovem saudável não consanguíneo Gravidez de termo Nascença peso de 32 Kg comprimento 50cm perímetro cefálico 35 cm Teste do pezinho com alterações (fim da primeira semana)
")
22
Antecedentes Familiares
Tia-avó paterna falecida aos 30 anos, sofria de atraso mental severo, com microcefalia Exame Objectivo Apirética Deficiente pigmentação da pele Cabelo claro Atraso psicomotor P cefálico abaixo do percentil 5 Ligeiro tremor das extremidades Sem hipertensão intercraneana Sem rigidez da nuca Fralda com cheiro particular Auscultação cardíaca e pulmonar s/ alterações aparentes

23
Hiperfenilalaninémia
Hipótese de diagnóstico Doença hereditária do metabolismo Hiperfenilalaninémia Exames complementares de diagnóstico Cromatografia de aminoácidos (HPLC) – Pico na região da Phe Urina – presença de ácido fenilpirúvico Estudos moleculares – Presença de mutação no gene PAH e LAT1 Actividade da fenilalanina hidrolase < 1% da encontrada em crianças na mesma idade Teste de Guthrie: fenilalanina (Phe) = 15mg/dl (N até 2mg/dl)
 – Pico na região da Phe. Urina – presença de ácido fenilpirúvico. Estudos moleculares – Presença de mutação no gene PAH e LAT1. Actividade da fenilalanina hidrolase < 1% da encontrada em crianças na mesma idade. Teste de Guthrie: fenilalanina (Phe) = 15mg/dl (N até 2mg/dl)")
24
Problemas Alterações da urina QI inferior a 50 Hiperactividade
Problemas de pele e cabelo Baixa esperança média de vida Possível invalidez permanente Sequelas irreversíveis do SN Convulsões

25
Hiperfenilalaninémia
1. A que se deve a elevação dos valores de fenilalanina sanguíneos neste caso? Hiperfenilalaninémia Mutação no gene PAH Acumulação de fenilalanina no sangue Défice de actividade da fenilalanina hidroxilase ou deficiência no co-factor.

26
2. Como se pode evitar as consequências deletérias desta alteração bioquímica?
fenilacetato Tentativa do organismo eliminar o excesso de finilalanina Acumulação de fenilalanina Fenilpiruvato fenilactato Dieta com alimentos pobres em fenilalanina. Complementar a alimentação com mistura de aminoácidos livres.

27
3. Acha que a tia-avó referida na história sofria da mesma doença que a criança em questão?
A PKU é uma doença autossómica recessiva cujos sintomas mais frequentes são: . Atraso mental num estado mais ou menos desenvolvido; . Microcefalia; . Convulsões; . Alterações da cor da pele olhos e cabelo; Sabendo que é uma doença autossómica recessiva, pelo que ambos os pais têm que ser portadores, associado aos sintomas que a tia-avó apresentava em vida constatamos que esta sofrera da mesma doença.

28
4. Estes doentes são frequentemente alourados, de cor clara, muitas vezes em contraste com a restante família. Porquê? Fenilalanina Tirosina DOPA Melanina Tirosinase PAH

29
5. Que dieta recomendaria a este doente e porquê?
Recomendaria uma dieta especial evitando comidas com alto teor de proteína como carnes vermelhas, ovos, aves, peixe, leite e queijo, e também o aspartame, um adoçante artificial. Isto porque todas as proteínas naturais contêm fenilalanina.

30
6. O que se deve esperar dos valores de tirosina plasmática na doença hereditária do metabolismo da fenilalanina mais frequente? Justifique. Devem ser esperados valores baixos de tirosina por défice da enzima fenilalanina hidroxilase que catalisa o processo de conversão da fenilalanina em tirosina.

31
Tratamento correcto da patologia
7. Quando são detectados níveis elevados de fenilalanina no sangue devem ser rastreadas outras doenças hereditárias do metabolismo dos AA, além da mais frequente. Qual a importância prática desta actuação? Há várias anomalias que podem causar uma hiperfenilalaninémia: Alterações na enzima fenilalanina hidroxilase (+ frequente) Défice do co-factor da fenilalanina hidroxilase 5,6,7,8-tetrahidrobiopterina por alterações na enzima dihidrobiopterina redutase ou na enzima dihidrobiopterina sintetase. Diagnóstico correcto da causa da hiperfenilalaninémia condiciona Diferentes objectivos do tratamento a ser administrado Tratamento correcto da patologia Diferente causa
 Défice do co-factor da fenilalanina hidroxilase 5,6,7,8-tetrahidrobiopterina por alterações na enzima dihidrobiopterina redutase ou na enzima dihidrobiopterina sintetase. Diagnóstico correcto da causa da hiperfenilalaninémia. condiciona. Diferentes objectivos do tratamento a ser administrado. Tratamento correcto da patologia. Diferente causa.")
32
Se a anomalia residir na fenilalanina hidroxilase. Tratamento:
-restrição dietética da fenilalanina, suplementada com tirosina, de modo a repor os valores normais de fenilalanina. Se a hiperfenilalaninémia for causada pela diminuição das concentrações do 5,6,7,8-tetrahidrobiopterina Tratamento : restauração da normal neurotransmissão monoaminérgica, já que a diminuição dos valores de tetrahidrobiopterina vão conduzir à disfunção da tirosina hidroxilase e da triptofano hidroxilase, uma vez que a tetrahidrobiopterina também é co-factor destas enzimas, cuja actividade é fundamental para a síntese de neurotransmissores como a serotonina e as catecolaminas. é necessária a administração de precursores tais como L-Dopa ou carbiopa e hidroxitriptofano. reposição dos valores normais de fenilalanina e tirosina como citado anteriormente ou então através da administração do próprio co-factor, para que sejam restituídas as reacções metabólicas normais;

33
8. Qual a importância desta alteração do metabolismo dos AA para o futuro do bebé?
Durante os primeiros meses de vida os bebes que nascem com fenilcetonúria parecem normais. Caso não iniciem o tratamento, entre os 3-6 meses começam a perder o interesse pelo meio que os rodeia, tornando-se evidente ao ano de idade um atraso no desenvolvimento psico-motor. Manifestam também outros sintomas da doença (convulsões, tremor das extremidades, cheiro particular da urina, pigmentação deficiente da pele e cabelo, entre outros). Caso não seja diagnosticada a tempo e tratada, o bebé poderá desenvolver atraso mental profundo que inclusivamente o levará à morte importância do rastreio desta doença em recém-nascidos através do “teste do pezinho”. Quando diagnosticada a doença, o seu tratamento condicionará toda a vida futura do bebé, inclusive a sua vida adulta, tendo de limitar a sua ingestão alimentar a alimentos que não possuam fenilalanina. Apesar desta situação por vezes se tornar um pouco incómoda na vida social, já que impõe determinadas restrições, é fundamental para que o indivíduo possa crescer e desenvolver-se normalmente, mantendo a sua integridade mental. Verificou-se num estudo que o Q.I. médio de fenilcetonúricos tratados em poucas semanas após o nascimento era 93, em contraste com um grupo de controlo que começou a ser tratado após o 1º ano de vida, com Q. I. médio 53. Precocidade do diagnóstico e tratamento: realização da triagem neonatal Prevenção de danos cerebrais irreversíveis. Diagnóstico e tratamento precoce e a existência de uma equipa multidisciplinar são portanto importantes para o futuro do bébé
. Caso não seja diagnosticada a tempo e tratada, o bebé poderá desenvolver atraso mental profundo que inclusivamente o levará à morte importância do rastreio desta doença em recém-nascidos através do teste do pezinho . Quando diagnosticada a doença, o seu tratamento condicionará toda a vida futura do bebé, inclusive a sua vida adulta, tendo de limitar a sua ingestão alimentar a alimentos que não possuam fenilalanina. Apesar desta situação por vezes se tornar um pouco incómoda na vida social, já que impõe determinadas restrições, é fundamental para que o indivíduo possa crescer e desenvolver-se normalmente, mantendo a sua integridade mental. Verificou-se num estudo que o Q.I. médio de fenilcetonúricos tratados em poucas semanas após o nascimento era 93, em contraste com um grupo de controlo que começou a ser tratado após o 1º ano de vida, com Q. I. médio 53. Precocidade do diagnóstico e tratamento: realização da triagem neonatal. Prevenção de danos cerebrais irreversíveis. Diagnóstico e tratamento precoce e a existência de uma equipa multidisciplinar são portanto importantes para o futuro do bébé.")
34
Referências http://www.elmhurst.edu/~chm/vchembook/images/635pku2.gif
Stryer, L.; Berg, J. M.; Tymoczko, J. L. (2002), Biochemistry (5th ed.), New York: W. H. Freeman, ISBN ; pp Harper's Illustrated Biochemistry (Harper's Biochemistry), 26th ed., 2003; pp Nelson, D. L. & Cox, M. M. (2000), Leningher Principles of Biochemistry, Worth Publishers, New York; pp
, Biochemistry (5th ed.), New York: W. H. Freeman, ISBN ; pp Harper s Illustrated Biochemistry (Harper s Biochemistry), 26th ed., 2003; pp Nelson, D. L. & Cox, M. M. (2000), Leningher Principles of Biochemistry, Worth Publishers, New York; pp")
Apresentações semelhantes













>")





