Carregar apresentação
This is a modal window.
1
Doença de Huntington Fernando Andersson Chemale
Guilherme Fagundes Bassols Marcelo Tonding Ferreira Rodrigo Sponchiado da Rocha

2
Introdução A coréia de Huntington é uma afecção degenerativa do sistema nervoso com padrão de herança autossômica dominante e penetrância completa; Os pacientes apresentam uma expansão da trinca CAG no gene IT15 do cromossomo 4; O quadro sindrômico caracteriza-se por movimentos involuntários coreiformes e alterações cognitivas, que se desenvolvem em torno dos 40 anos de idade, progredindo até a morte em um período de aproximadamente 15 anos;

3
Histórico George Huntington primeira descrição Coréia hereditária
The Medical and Surgical Reporter

4
A doença comumente se inicia por leves abalos dos músculos da face, que aumentam gradativamente em violência e variedade. As pálpebras são mantidas piscando, a testa franzida depois elevada, o nariz torcido para um lado e depois para outro e a boca se volta em direções variadas, dando ao paciente a aparência mais ridícula que se possa imaginar. Parece haver alguma força oculta, algo que de certa forma está brincando com a vontade e de algum modo dificultando e pervertendo os seus desígnios; e depois que a vontade pára de exercer sua força, assume o controle e mantém a pobre vítima numa agitação contínua enquanto ela permanece acordada.

6
Particularidades Natureza hereditária; Tendência à insanidade;
Idade tardia de aparecimento;

7
Histórico T. Meynert em 1877 demonstrou que os sintomas da DH estão associadas com alterações no núcleo caudado; Anton em 1896 e Alzheimer em 1911 demonstraram que a DH atinge todo o estriado que corresponde ao núcleo caudado e putâmen

8
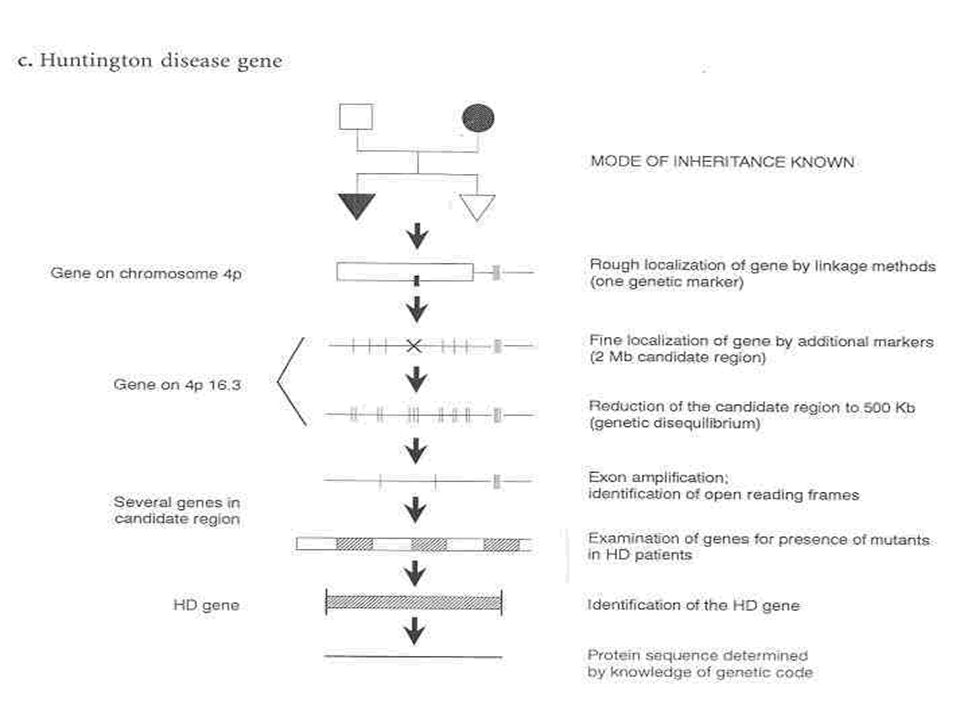
Gusella, em 1983, conseguiu localizar o gene responsável pela síndrome de Huntington;
Em 1993, o ‘Huntington Disease Collaborative Research Group’ descobriu que a alteração gênica responsável pela DH era a expansão da repetição dos trínucleotídeos CAG localizados na extremidade 5’ do gene IT15 no braço curto do cromossomo 4.

9
Epidemiologia 5 a 10 por 100.000 indivíduos
Regiões de alta prevalência: Tasmânia e lago Maracaibo na Venezuela Regiões de baixa prevalência: Japão, China, Finlândia

10
GENÉTICA

11
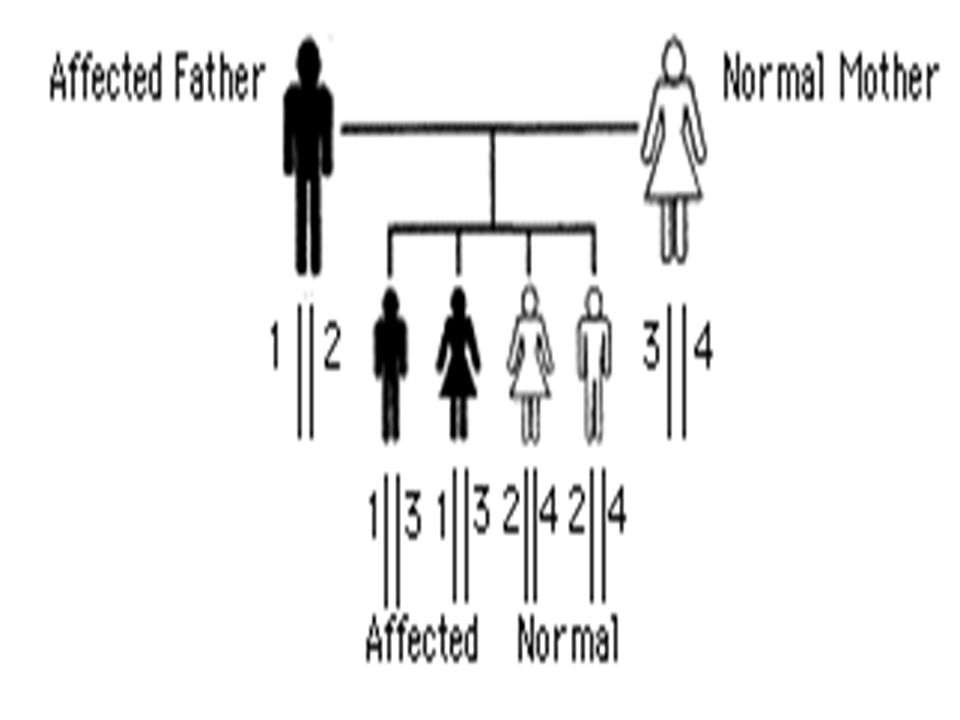
Tipo de herança Autossômica dominante, com penetrância completa;
Todo indivíduo afetado possui um genitor afetado; Distribuição igual entre homens e mulheres; Todo indivíduo afetado transmite a característica para metade de sua prole;

13
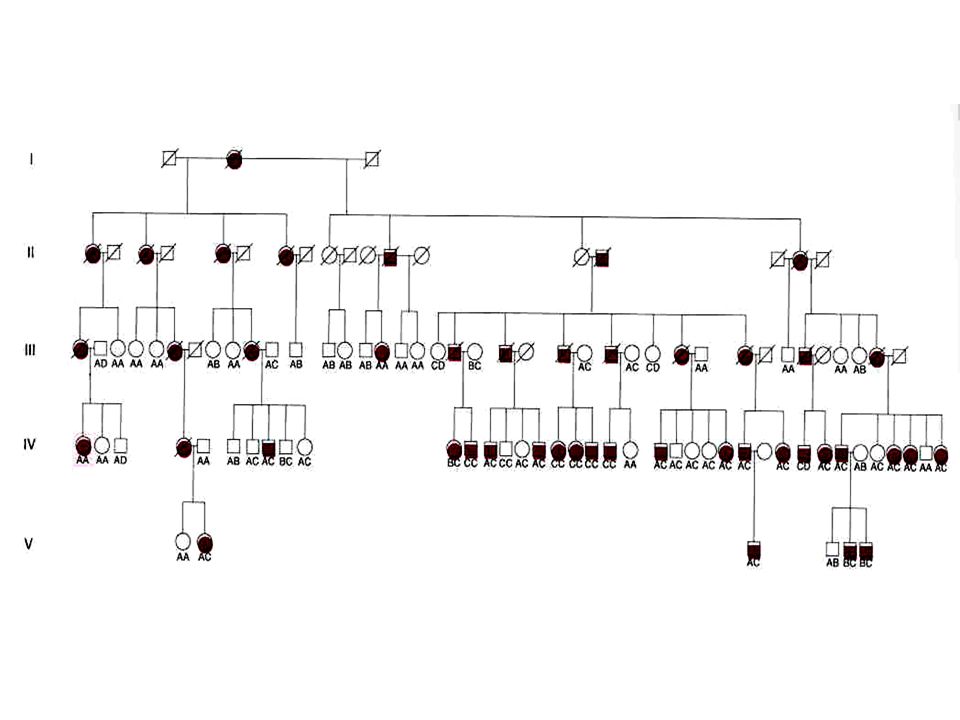
Homozigotos X Heterozigotos
Expressão fenotípica idêntica; Lago Maracaibo (Venezuela): Homozigotos e heterozigotos; “Efeito do fundador”
: Homozigotos e heterozigotos; Efeito do fundador")
15
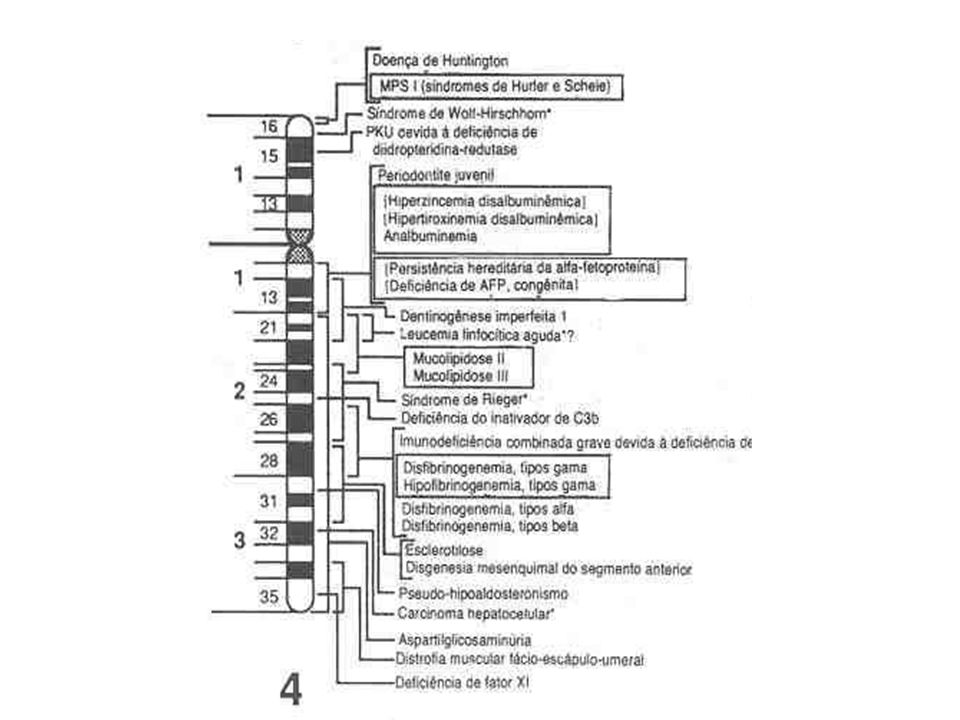
Cromossomo 4

17
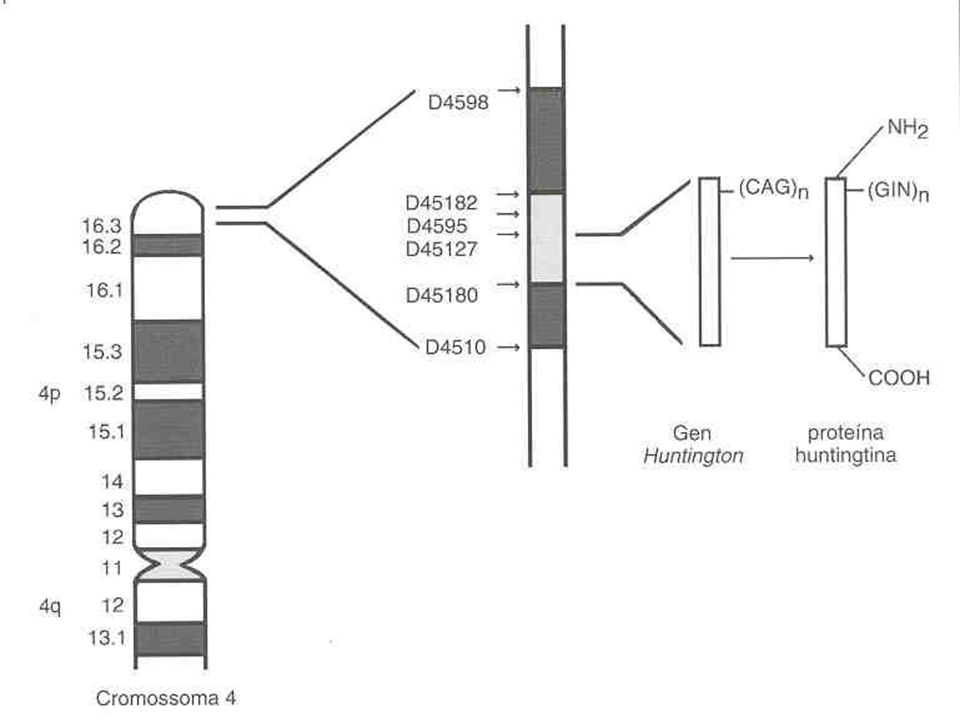
Interesting Transcript 15 (IT15)
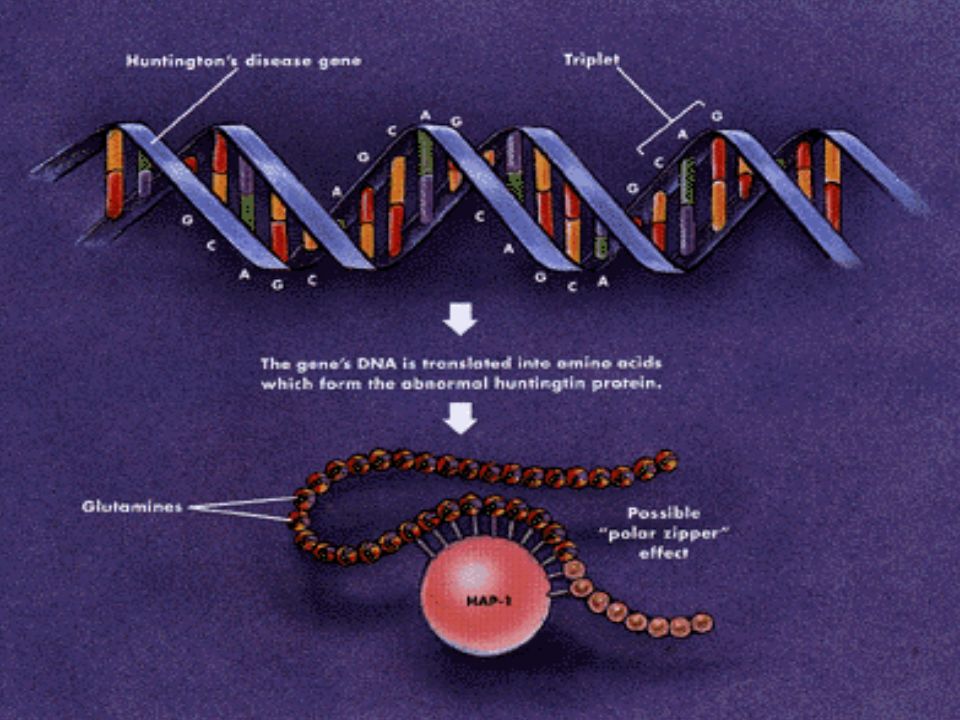
O gene mutante foi localizado na extremidade telomérica do braço curto na região 4p16.3 do cromossomo 4; Extremidade 5’ Múltiplas cópias do trinucleotídeo CAG em tandem; CAG Glutamina; Repetições CAG Poliglutamina;

18
Gen IT15 Huntingtina

20
Extremidade telomérica do gene IT15 Repetições do trinucleotídeo CAG
A Mutação do Gene IT15 Extremidade telomérica do gene IT15 Repetições do trinucleotídeo CAG

21
Gene IT15 normal Até 35 repetições de trinucleotídeo CAG
Mutação do gene IT15 Gene IT15 normal Até 35 repetições de trinucleotídeo CAG

22
Genótipo intermediário 35 a 40 repetições CAG
Mutação do gene IT15 Genótipo intermediário 35 a 40 repetições CAG

23
Doença de Huntigton Gene mutante Acima de 40 repetições CAG
Mutação do gene IT15 Doença de Huntigton Gene mutante Acima de 40 repetições CAG

25
Instabilidade das repetições na gametogênese
Herança materna Ovulogênese Aumento ou diminuição de 3 a 4 CAG Herança paterna Espermatogênese Duplicação do número de CAG

26
Antecipação Genética na DH
Prole com idade precoce de manifestação e/ou uma expressão mais grave da doença; Herança paterna DH juvenil

27
Polimorfismos adjacentes à repetição CAG
A deleção de 3 nucleotídeos adjacentes ao CAG tem correlação com diminuição na idade de início da doença; 96% dos pacientes com DH apresentaram repetições de 7 trinucleotídios CCG adjacentes à repetição CAG, não havendo influência fenotípica;

28
Mutações novas Evento raríssimo Baixa taxa mutacional
Suspeita de Investigar o genótipo mutação nova dos pais

29
Repetições expandidas instáveis
Fora da região codificadora Síndrome do X frágil (CGG e GCC) e distrofia miotônica Interferem na transcrição Dentro da região codificadora Doença de Huntington, AEC1, ADRPL, doença de Machado-Joseph, AMEB Não interferem na transcrição
 e distrofia miotônica. Interferem na transcrição. Dentro da região codificadora. Doença de Huntington, AEC1, ADRPL, doença de Machado-Joseph, AMEB. Não interferem na transcrição.")
30
FISIOPATOGENIA

31
HUNTINGTINA Proteína codificada pelo gene IT15 normal;
presente em vários tecidos do corpo, embora esteja concentrada no cérebro; a Huntingtina normal é encontrada quase exclusivamente no citoplasma;

32
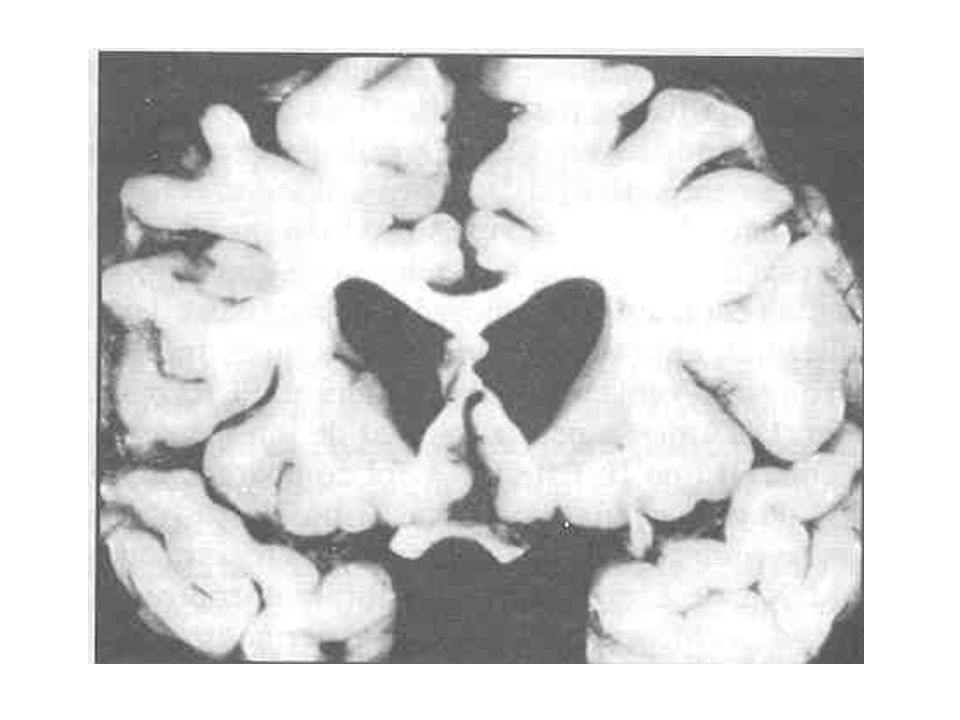
Huntingtina Tamminga et cols : no citoplasma a huntingtina estaria associada em parte com as membranas das vesículas do C. de Golgi Migração vesicular e exocitose de substâncias ( Neurotransmissores, enzimas,...)
")
33
Huntingtina Também é creditado a huntingtina um papel no mecanismo de apoptose celular, sugerindo que ela se ligaria a outras proteínas neuronais, formando complexos citotóxicos

34
HUNTINGTINA MUTANTE Na DH a huntingtina possui uma cadeia anormal de poliglutaminas que confere à sua estrutura novas propriedades que desencadeiam interações anômalas com outras proteínas; A huntingtina mutante encontra-se predominantemente no núcleo da célula, em comparação à huntingtina normal, que se encontra no citoplasma;

35
não há aumento nem diminuição da expressão do RNA huntington em neurônios dos núcleos caudado e putâmen, locais de maior severidade da DH Existência de outros fatores que interagem com a huntingtina, fazendo com que as células dos núcleos da base sejam mais vulneráveis proteínas HAP1, GAPDH,...

37
Huntingtina mutante Portanto, a interação mais intensa da huntingtina anormal com determinadas proteínas do cérebro causaria uma alteração ainda incerta no mecanismo de apoptose e na exocitose de substâncias pelos neurônios, causando progressiva degeneração dos núcleos da base e conseqüente atrofia cerebral

39
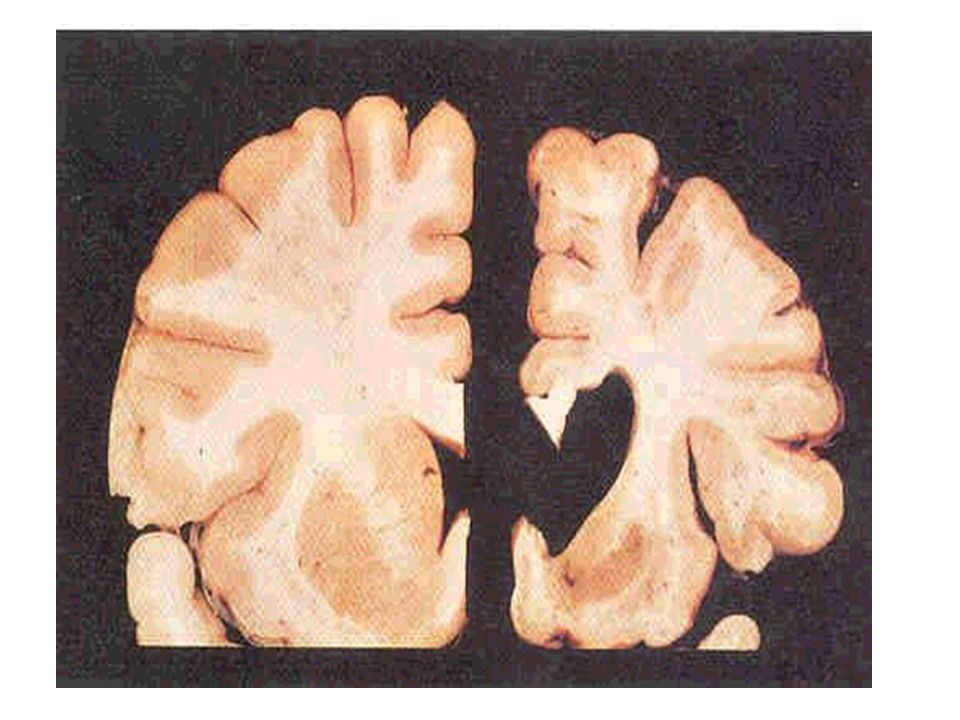
Núcleos da base

40
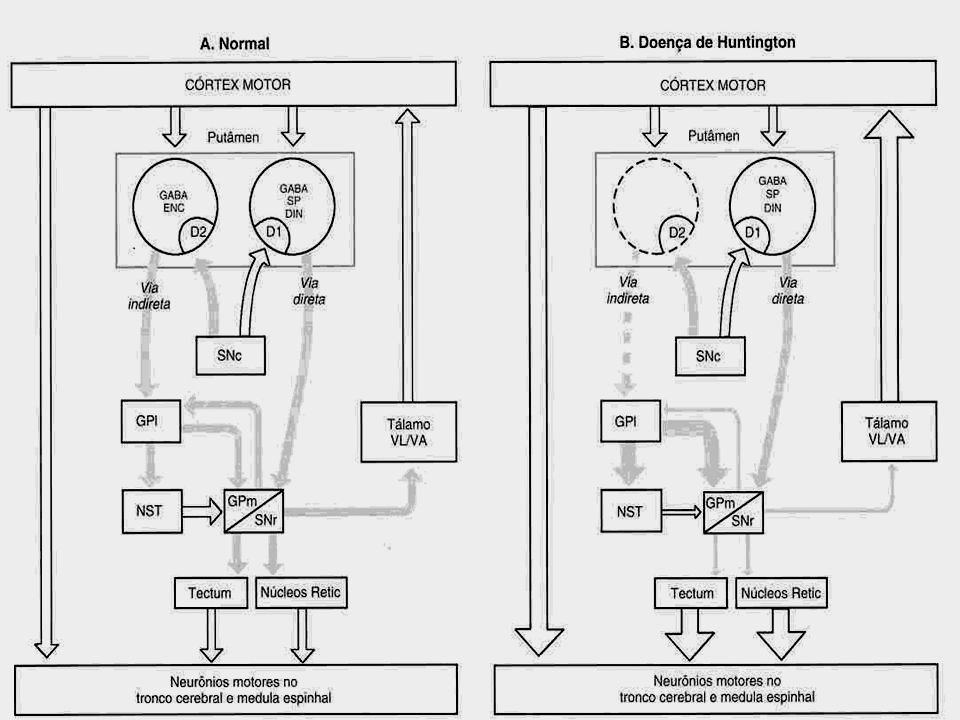
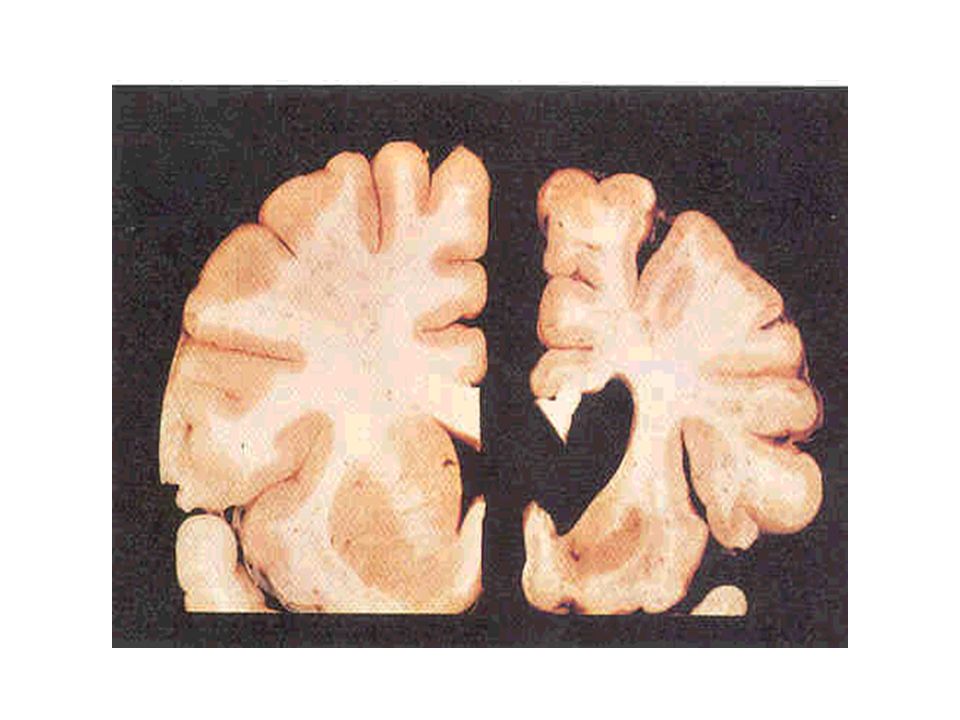
A modalidade de comprometimento do tecido cerebral na DH apresenta potencial capacidade de depletar substâncias próprias dos neurônios, como GABA, encefalinas e substância P. Há perda principalmente dos neurônios gabaérgicos do caudado e putâmen (Estriado).
..")
42
Mecanismo da coréia Perda da maioria dos corpos dos neurônios do caudado e putâmen e dos neurônios secretores de Ach de várias áreas do cérebro Perda da inibição do globo pálido e da substância negra descargas espontâneas de atividade do globo pálido e da substância negra MOVIMENTOS DE DISTORÇÃO

43
DEMÊNCIA Perda dos neurônios secretores de acetilcolina (Ach), especialmente os localizados nas áreas de pensamento do córtex cerebral
, especialmente os localizados nas áreas de pensamento do córtex cerebral.")
44
MANIFESTAÇÕES CLÍNICAS

45
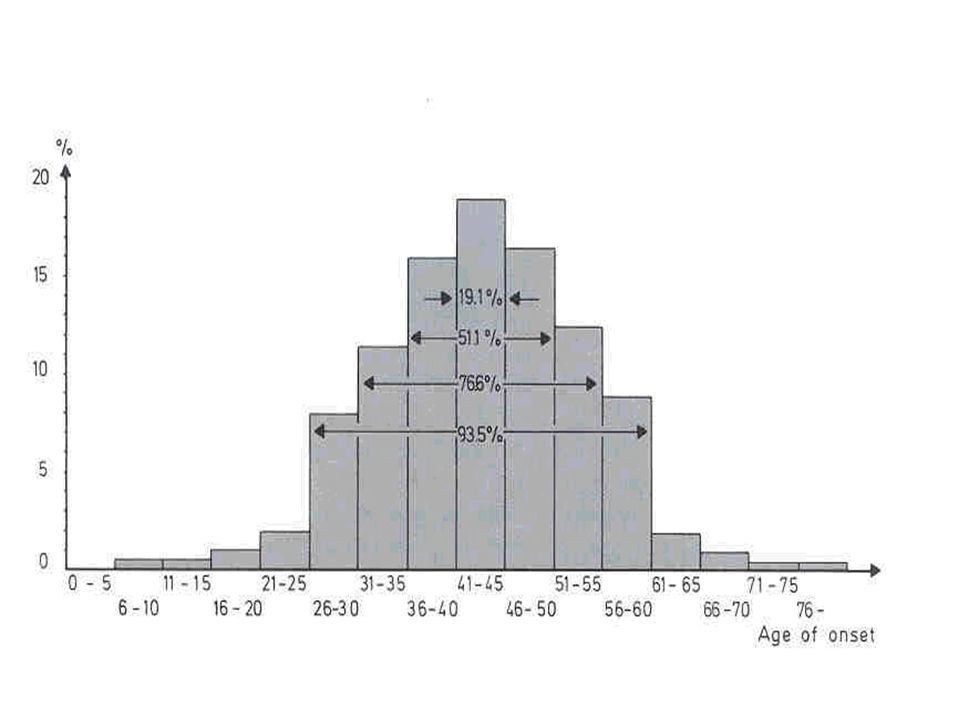
Idade atrasada de manifestação
A DH apresenta sintomas que em geral não são vistos antes dos 30 anos; Seleção natural Progenitores passam o gene para sua prole mesmo sem saber que são carreadores da mutação; Herança paterna Fator de risco para DH juvenil.

47
Manifestações clínicas
Distúrbios motores progressivos, com comprometimento dos movimentos voluntários e aparecimento de movimentos involuntários; Declínio da capacidade cognitiva global e presença de transtornos psiquiátricos

48
Manifestações motoras

49
Coréia É o sinal motor mais notável da doença, estando presente em cerca de 90% dos afetados; As manifestações coreiformes na face são representadas por contrações da bochecha, franzimento das sobrancelhas, movimentos labiais e nistagmo; Pode haver envolvimento do pescoço;

50
Coréia Nos membros, há envolvimento de múltiplas articulações;
Num estágio avançado pode haver o aparecimento de movimentos semelhantes ao balismo; Os movimentos estão continuamente presentes; Em geral, a coréia inicia-se distalmente e, com a evolução, torna-se generalizada, podendo interromper os movimentos voluntários.

52
Distonia Presente em 95,2% dos pacientes clinicamente sintomáticos;
É caracterizada por movimentos lentos anormais e por alterações posturais, tornando-se proeminente apenas no estágio final das doença;

53
Distúrbios oculomotores
São observados na maioria dos pacientes, estando presentes no estágio inicial da doença; A lentidão dos movimentos sacádicos está presente em 75% dos doentes, sendo os movimentos verticais mais afetados que os horizontais;

54
Bradicinesia e rigidez
São infreqüentes nas fases iniciais da doença. Entretanto, gradualmente, essas manifestações podem dominar no estádio final da doença, no qual o paciente torna-se severamente rígido e acinético;

55
Alterações na marcha Os distúrbios da marcha tornam-se mais pronunciados com a progressão da doença; Os pacientes podem apresentar quedas freqüentes.

56
Discinesia A perda da motricidade voluntária progride, com o curso da doença, até atingir a incapacidade de efetuar qualquer movimento proposto.

57
Alterações da fala A disartria, lentidão e a falta de iniciativa perturbam a fluência e a fala espontânea. Entretanto, a estrutura semântica, o vocabulário e a compreensão do discurso estão preservados até os estágios tardios da doença.

58
Disfagia A asfixia e a aspiração decorrentes da disfagia são causas comuns de morbidade Ocorre tardiamente na DH

59
Reflexos hiperativos Ocorrem precocemente em até 90% dos pacientes;
A resposta plantar em extensão é predominante nos casos juvenis ou em adultos em casos avançados da doença;

60
Incontinência urinária e fecal
Raras entre os doentes recentemente diagnosticados; Acometem 20% dos pacientes em fase terminal;

61
Manifestações psiquiátricas e cognitivas

62
Diminuição da memória Freqüente redução da capacidade de memorização, inicialmente afetando a memória visual, espacial e auditiva; A memória verbal é secundariamente afetada; A orientação em tempo e espaço é preservada até as fases finais da doença;

63
Déficit de atenção e concentração
Presentes no estágio inicial da doença, e estão relacionados com alteração da percepção visual dos pacientes;

64
Demência É o sintoma inicial em 10% dos casos;
90% dos pacientes desenvolvem durante o curso da doença; A lentidão do pensamento pode ser observada precocemente;

65
Mudanças no humor e afeto
A depressão é o principal sintoma psiquiátrico e acomete 40% dos doentes; Maior taxa de suicídio; Distúrbios de conduta: apatia, ansiedade, agressividade, desinibição sexual e alcoolismo;

66
Cerca de 50% dos pacientes com DH em estágio avançado apresentam pensamentos ilusórios;
Na fase terminal, podem ser observados distúrbios do sono e inversão do ritmo circadiano;

67
DH juvenil 5,4% dos casos; Manifesta-se inicialmente com parkinsonismo progressivo, demência e convulsões, sendo menos freqüente a coréia, diferentemente da forma adulta; Deterioramento clínico mais acelerado;

68
Relação genótipo/fenótipo
O número de repetições CAG está diretamente relacionado a rapidez da deterioração clínica do paciente, e inversamente associado à idade de aparecimento da doença; Não parece haver nenhuma relação entre o número de repetições CAG e o tipo de sintomas (motores ou comportamentais) iniciais da DH;
 iniciais da DH;")
70
DIAGNÓSTICO

71
Diagnóstico Clínico Imagético Genético

72
Diagnóstico clínico Disfunções motoras Movimentos coreicos;
Distúrbios cognitivos; História familiar positiva;

73
Diagnóstico imagético
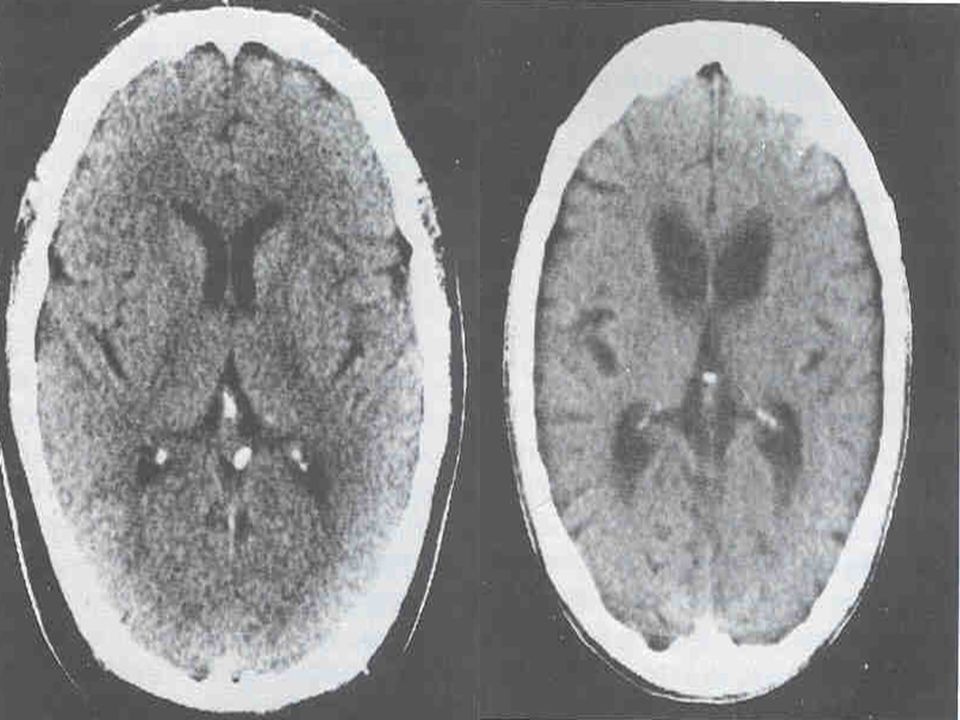
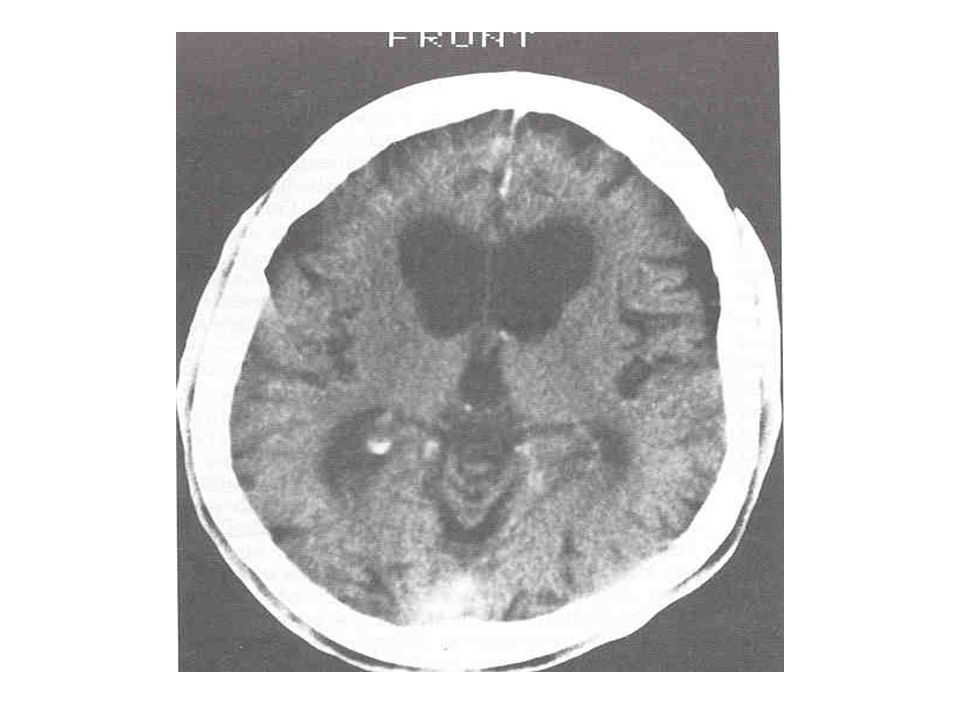
Alterações básicas Atrofia do estriado; Atrofia cortical TC = RNM; RNM permite medidas volumétricas;

80
Diagnóstico genético Confirmação da doença de Huntington;
Melhor método para realizar diagnóstico diferencial; Implicações médicas, psicológicas, éticas e financeiras;

81
Técnica DNA isolado de linfócitos periféricos;
Amplificação utilizando a reação da polimerase em cadeia (PCR); Eletroforese; Hibridização com sonda (CAG)15 ; Contagem do número de repetições CAG; Menos de Mais de 40 Repetições CAG Repetições CAG Normal DH
; Eletroforese; Hibridização com sonda (CAG)15 ; Contagem do número de repetições CAG; Menos de 35 Mais de 40. Repetições CAG Repetições CAG. Normal DH.")
82
Teste genético para pacientes sintomáticos
Sintomas neurológicos compatíveis com DH; Diagnóstico diferencial de outras doenças que se apresentam com manifestações coreiformes; Se o paciente apresentar a expansão do trinucleotídeo e não tiver história familiar positiva Investigar paternidade

83
Teste genético em pacientes assintomáticos
Filhos de pais portadores de DH; 50% de chance de desenvolver a doença; Perspectivas de trabalho, planejamento pessoal e familiar; Alterações psiquiátricas ao saber que irá desenvolver uma doença assustadora que evoluirá para o óbito;

84
Aconselhamento genético
Proposto pela “International Huntington Association” e pela “World Federation of Neurology”. “Guidelines” sobre as recomendações que devem ser dadas aos pacientes que procuram os laboratórios de genética. Recomendações baseadas nos princípios éticos, no conhecimento da DH e nas técnicas de genética molecular.

85
Aconselhamento genético
1) Todo indivíduo que quiser realizar o teste deve receber informações atualizadas e relevantes, para dar seu consentimento voluntário. 2) Fazer o teste é uma decisão única e exclusiva do indivíduo interessado. Não devem ser consideradas solicitações de terceiros. 3) O participante deve ser encorajado a escolher uma pessoa para acompanhá-lo em todas as etapas do teste. 4) O teste e o aconelhamento devem ser realizados em unidades especializadas, preferencialmente em um departamento universitário.
 Todo indivíduo que quiser realizar o teste deve receber informações atualizadas e relevantes, para dar seu consentimento voluntário. 2) Fazer o teste é uma decisão única e exclusiva do indivíduo interessado. Não devem ser consideradas solicitações de terceiros. 3) O participante deve ser encorajado a escolher uma pessoa para acompanhá-lo em todas as etapas do teste. 4) O teste e o aconelhamento devem ser realizados em unidades especializadas, preferencialmente em um departamento universitário.")
86
Diagnóstico pré-natal
Cordocentese Amniocentese Análise de vilosidades coriônicas Muitos centros de genética não realizam, pois além de ser um método invasivo, causa um grande impacto nos pais e não implica em nenhuma estratégia terapêutica. Tais centros preferem apenas o aconselhamento.

87
Diagnóstico diferencial
Grupos em que o diagnóstico é difícil: Pacientes com quadro clínico de coréia, mas sem história familiar positiva; Pacientes com risco de desenvolver coréia e que se encontram com sintomas psiquiátricos, mas sem coréia; Pacientes com coréia provocada por outra doença;

88
Diagnóstico diferencial
Neuroacantocitose: Provoca movimentos involuntários, demência e atrofia do caudado. Provocada por defeito nas membranas plasmáticas. Coréia de Sydenhan: Manifestação tardia da febre reumática. Confunde-se com DH juvenil. Manejo e prgnóstico diferentes.

89
Diagnóstico diferencial
Hipertireoidismo Coréia de Huntington Lúpus eritematoso sistêmico Neurosífilis Doença de Wilson Encefalite Infartos dos gânglios da base Discinesias tardias provocadas por fármacos

90
Tratamento A DH é uma enfermidade incurável;
O tratamento é puramente sintomático, com drogas bloqueadoras dos receptores dopaminérgicos (fenotiazinas ou Haloperidol); Esses fármacos podem induzir um quadro de discinesia tardia superposta ao distúrbio crônico
; Esses fármacos podem induzir um quadro de discinesia tardia superposta ao distúrbio crônico.")
91
Conclusão A doença de Huntington é a mais comum doença neurodegenerativa hereditária. Os mecanismos moleculares envolvidos na gênese desse distúrbio ainda não foram suficientemente esclarecidos. As manifestações clínicas da DH são severas e comprometem dramaticamente a qualidade de vida dos doentes. O diagnóstico genético, por sua vez, envolve aspectos complexos que devem ser analisados e discutidos tanto com o doente quanto com os seus familiares. Por fim, é imperiosa a necessidade uma atitude efetiva da equipe médica com o objetivo de otimizar o diagnóstico e manejo de pacientes com DH.