Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Púrpuras Trombocitopênicas
Trombocitopenia: contagem de plaquetas abaixo de /µl Púrpura: Manchas avermelhadas causadas pelo extravasamento de hemácias na derme que não desaparecem à vitropressão. Inicialmente são mais avermelhadas e posteriormente se tornam acastanhadas ou marrom-claro.

3
Púrpuras Trombocitopênicas
Manifestações clínicas: Hemorrágicas: petéquias, equimoses e sangramento de mucosas. Adicionais : Esplenomegalia, adenomegalia, lesões cutâneas ou articulares.

4
Púrpuras Trombocitopênicas Hereditárias X Adquiridas
95% das TROMBOCITOPENIAS ISOLADAS são autoimunes ou induzidas por medicamentos ; Muitos casos de PT Congênita podem ser erroneamente diagnosticados como PTI ; Novos métodos de detecção de defeitos moleculares

5
Púrpuras Trombocitopênicas
Hereditárias Em todas as formas hereditárias há distúrbio da plaquetogênese na MO; Sobrevida das plaquetas normal ou diminuída; A lesão básica em todas as hereditárias: Hipoplasia ou falta de maturação normal dos megacariócitos medulares; Podem exibir defeitos funcionais.

6
Quando suspeitar de PT Hereditária ?
Início ao nascimento; História familiar de trombocitopenia; 2 ou mais parentes Tios ou primos maternos Sangramento desproporcional à contagem plaquetária; Alterações clínicas associadas: Ausência de rádio, retardo mental, insuficiência renal, déficit auditivo, catarata, leucemia, úvula bífida, arco aórtico direito, hematoquezia neonatal, eczema, infecções frequentes, disfunção plaquetária

7
Quando suspeitar de PT Hereditária?
Níveis de trombocitopenia estáveis durante anos Alterações no esfregaço de sangue periférico : Plaquetas de tamanhos anormais (pequenas, grandes - macroplaquetas, gigantes); Ausência de grânulos alfa (Sínd. das Plaquetas Cinzentas); Corpúsculos de Dohle-like (MYH9) Falha de resposta à terapia para PTI Dificuldade em diferenciar pela resposta hematológica a PTI Refratária da PT Hereditária Algumas PT Hereditária respondem a IVIG, corticosteróide e/ou esplenectomia
; Ausência de grânulos alfa (Sínd. das Plaquetas Cinzentas); Corpúsculos de Dohle-like (MYH9) Falha de resposta à terapia para PTI. Dificuldade em diferenciar pela resposta hematológica a PTI Refratária da PT Hereditária. Algumas PT Hereditária respondem a IVIG, corticosteróide e/ou esplenectomia.")
8
Como classificar as PT Hereditárias?
Localização do defeito molecular; Mecanismo: produção baixa ou destruição acelerada; Tamanho das plaquetas no esfegaço: muito grandes, normais ou pequenas. Outros achados – microcitose RBC, Dohle-like bodies em neutrófilos Modo de transmissão: autossômica dominante, recessiva ou ligada ao X; Alterações clínicas e laboratoriais associadas: Ausência de rádio, IR, déficit auditivo; Anormalidades laboratoriais – citometria de fluxo para glicoproteínas de plaquetas, teste de função plaquetária

9
Trombocitopenias congênitas específicas
Síndrome de Wiskott-Aldrich; Trombocitopenia ligada ao X; Disfunção plaquetária familiar com predisposição à leucemia aguda; Trombocitopenia amegacariocítica; Trombocitopenia amegacariocítica com sinostose rádio-ulnar; Trombocitopenia com ausência de rádio (TAR = Thrombocitopenia with absent radio); Síndrome de Bernard-Soulier; Síndrome DiGeorge;
; Síndrome de Bernard-Soulier; Síndrome DiGeorge;")
10
Trombocitopenias congênitas específicas
Doença de Von Willebrand tipo 2B – Tipo plaquetário da doença de Von Willebrand Macrotrombocitopenia benigna do mediterrâneo; Trombocitopenia ligada ao X e diseritropoiese com ou sem anemia; Trombocitopenia-talassemia ligada ao X; Doença relacionada ao MYH9 Anomalia de May-Hegglin Síndrome de Fechtner Síndrome de Sebastian Síndrome de Epstein Síndrome da plaqueta cinza

11
PÚRPURAS PLAQUETÁRIAS TROMBOCITOPÊNICAS ADQUIRIDAS HIPERESPLENISMO
APLASIA MEGACARIOCÍTICA INFECÇÃO HIV ÁLCOOL DEFICIÊNCIA DE VIT B12 DEFICIÊNCIA DE FÓLICO DEFICIÊNCIA DE FERRO FALTA DE PRODUÇÃO DISTRIBUIÇÃO IRREGULAR ADQUIRIDAS HIPERESPLENISMO HEPATOPATIA ESQUISTOSSOMOSE

12
PÚRPURAS PLAQUETÁRIAS TROMBOCITOPÊNICAS ADQUIRIDAS CAUSA IMUNOLÓGICA
PTI TROMBOCITOPENIA CÍCLICA DROGAS PÚRPURA ALO-IMUNE NEONATAL PÓS TRANSFUSÃO DESTRUIÇÃO PERIFÉRICA FALTA DE PRODUÇÃO DISTRIBUIÇÃO IRREGULAR ADQUIRIDAS PLT FAGÓCITO Comple mento Ac

13
PÚRPURAS PLAQUETÁRIAS TROMBOCITOPÊNICAS ADQUIRIDAS CAUSA IMUNOLÓGICA
PTI TROMBOCITOPENIA CÍCLICA DROGAS PÚRPURA ALO-IMUNE NEONATAL PÓS TRANSFUSÃO DESTRUIÇÃO PERIFÉRICA FALTA DE PRODUÇÃO DISTRIBUIÇÃO IRREGULAR ADQUIRIDAS FAGÓCITO

14
PÚRPURAS PLAQUETÁRIAS TROMBOCITOPÊNICAS ADQUIRIDAS CAUSA IMUNOLÓGICA
PTI TROMBOCITOPENIA CÍCLICA DROGAS PÚRPURA ALO-IMUNE NEONATAL PÓS TRANSFUSÃO DESTRUIÇÃO PERIFÉRICA FALTA DE PRODUÇÃO DISTRIBUIÇÃO IRREGULAR ADQUIRIDAS FAGÓCITO

15
PÚRPURAS PLAQUETÁRIAS TROMBOCITOPÊNICAS ADQUIRIDAS
DROGAS ASSOCIADAS À PTI ÁLCOOL ACETAMINOFEN ALFA-METILDOPA CARMABAZEPINA CLORTALIDONA CLOROTIAZINA CIMETIDINA COCAÍNA DIGITAL DIFENIL HIDANTOÍNA FUROSEMIDA HEPARINA HEROÍNA INTERFERON INDOMETACINA PENICILINA PROCAINAMIDA QUINIDINA RANITIDINA RIFAMPICINA SULFONAMIDAS DROGAS ASSOCIADAS À PTI ÁLCOOL ACETAMINOFEN ALFA-METILDOPA CARMABAZEPINA CLORTALIDONA CLOROTIAZINA CIMETIDINA COCAÍNA DIGITAL DIFENIL HIDANTOÍNA FUROSEMIDA HEPARINA HEROÍNA INTERFERON INDOMETACINA PENICILINA PROCAINAMIDA QUINIDINA RANITIDINA RIFAMPICINA SULFONAMIDAS CAUSA IMUNOLÓGICA PTI TROMBOCITOPENIA CÍCLICA DROGAS PÚRPURA ALO-IMUNE NEONATAL PÓS TRANSFUSÃO DESTRUIÇÃO PERIFÉRICA FALTA DE PRODUÇÃO DISTRIBUIÇÃO IRREGULAR ADQUIRIDAS CAUSA NÃO IMUNOLÓGICA PTT / SHU INFECÇÕES DROGAS NEOPLASIA GRAVIDEZ DAI

16
Púrpuras Trombocitopênicas Idiopáticas
Trombocitopenia < /mm3 Encurtamento da sobrevida plaquetária Presença de Anticorpos Anti-plaquetários no plasma e/ou plaquetas AUMENTO DE MEGACARIÓCITOS NA MEDULA ÓSSEA Difícil avaliação na prática clínica Critério mais utilizado, associado à clínica. Ausência de outros sinais ao exame físico, exceto a síndrome purpúrica DIAGNÓSTICO DE EXCLUSÃO

17
PTI Aguda versus Crônica
O que define se a PTI é crônica ou não é a duração maior ou menor que 6 meses Aguda: Entre 2 a 8 anos Em 80% dos casos precedida de infecção viral Associação com doenças auto-imunes rara Eosinofilia e linfocitose podem ocorrer Duração: de 2-6 semanas Evolução: Remissão espontânea em 85% dos casos Crônica: Mais freqüente em adultos Raramente precedida de infecção viral Associação com doenças auto-imunes comum Eosinofilia e linfocitose raramente Duração: maior que 6 meses Evolução: curso flutuante e crônico

18
Fatores desencadeantes
Infecções Virais 1 a 2 semanas antes da apresentação clínica (PTI) PT Imune Secundária : -Vacinação -Medicamentos (Ex: Xaropes com Guaiacolato – Veículo comum em antitussígenos, AINEs, rifampicina,penicilinas, anfotericina B, furosemida, carbamazepina, valproato, hipoglicemiantes orais, benzodiazepínicos, heparina, alfa-metil-dopa ). -Doenças auto-imunes (LES, Síndrome de Evans, Tireoidite de Hashimoto) -Neoplasias (LLC ou linfomas de baixa agressividade em adultos) Há um grupo de pacientes em que não se identifica o fator desencadeante Pós-transfusional
 PT Imune Secundária : -Vacinação. -Medicamentos (Ex: Xaropes com Guaiacolato – Veículo comum em antitussígenos, AINEs, rifampicina,penicilinas, anfotericina B, furosemida, carbamazepina, valproato, hipoglicemiantes orais, benzodiazepínicos, heparina, alfa-metil-dopa ). -Doenças auto-imunes (LES, Síndrome de Evans, Tireoidite de Hashimoto) -Neoplasias (LLC ou linfomas de baixa agressividade em adultos) Há um grupo de pacientes em que não se identifica o fator desencadeante. Pós-transfusional.")
22
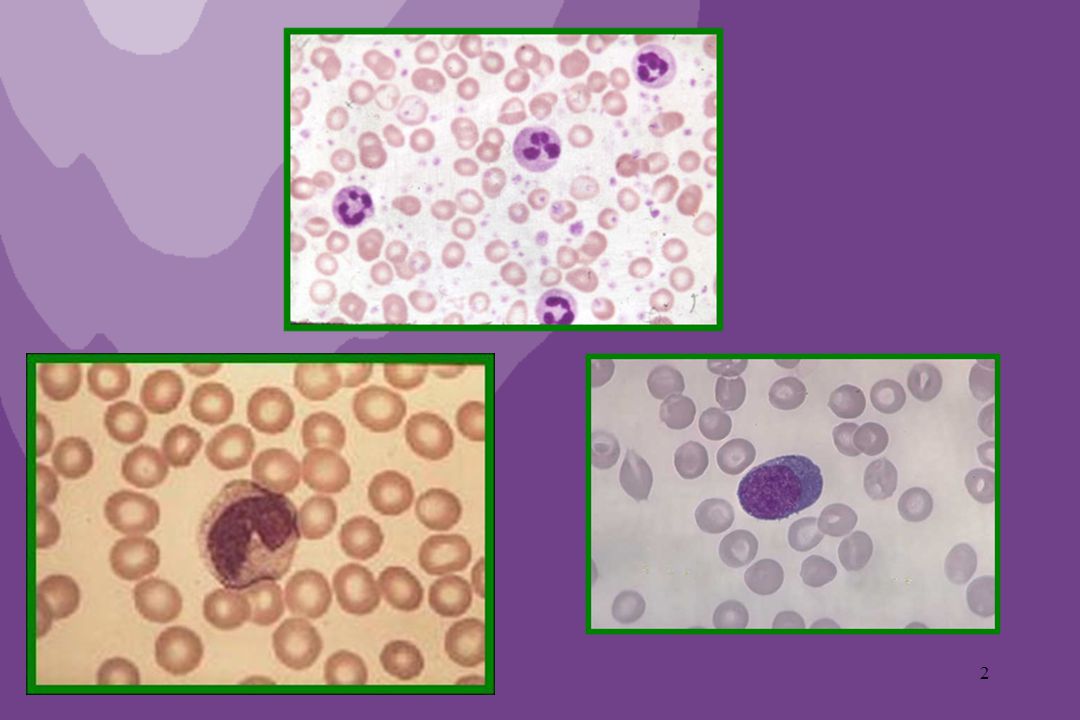
Setor megacariocítico hiperplástico

24
Tratamento inicial recomendado
Iniciar prednisona (1mg/kg) e caso ocorra resposta diminuir a dose devagar mantendo uma satisfatória contagem de plaquetas e efeitos colaterais toleráveis. Em pacientes Rh+ que não responderam bem a prednisona e/ou suas doses não podem ser diminuídas para níveis seguros deve-se administrar anti-D. Entretanto, quando a contagem cair abaixo de /μL esse abordagem deve ser mantida por 6 meses e depois eventualmente suspender o tratamento. IgIV, anti-D e esteróides são usados antes da cirurgia para elevar as plaquetas para níveis seguros. Danazol. Dose de 200mg é dada inicialmente com a dose total de prednisona. A resposta ocorre lentamente e a terapia deve ser continuada por 3-6m antes de abandoná-la. Em pacientes responsivos, a dose da prednisona deve ser diminuída lentamente e se possível interrompida, enquanto dá continuoidade ao danazol em doses plenas por no minímo 1 ano quando se começa a diminuir lentamente a dose.
 e caso ocorra resposta diminuir a dose devagar mantendo uma satisfatória contagem de plaquetas e efeitos colaterais toleráveis. Em pacientes Rh+ que não responderam bem a prednisona e/ou suas doses não podem ser diminuídas para níveis seguros deve-se administrar anti-D. Entretanto, quando a contagem cair abaixo de /μL esse abordagem deve ser mantida por 6 meses e depois eventualmente suspender o tratamento. IgIV, anti-D e esteróides são usados antes da cirurgia para elevar as plaquetas para níveis seguros. Danazol. Dose de 200mg é dada inicialmente com a dose total de prednisona. A resposta ocorre lentamente e a terapia deve ser continuada por 3-6m antes de abandoná-la. Em pacientes responsivos, a dose da prednisona deve ser diminuída lentamente e se possível interrompida, enquanto dá continuoidade ao danazol em doses plenas por no minímo 1 ano quando se começa a diminuir lentamente a dose.")
25
Tratamento corticóide Imunoglobulina IV Célula B Macrófago plaqueta
Receptor IIb/IIIa – receptor de fibrinogênio Receptor da porção Fc da Imunoglobulina

26
Púrpura trombocitopênica imunológica -
Esplenectomia É recomendada nas seguintes situações: Níveis seguros de plaquetas não podem ser mantidos; Remissão é considerada improvável; Severa toxicidade por drogas; Abordagem terapêutica muito difícil para o paciente suportar. 75%-85% dos pacientes tem uma resposta inicial, desses 25-40% apresentam recaída entre 5-10 anos.

27
PTI- tratamento de pacientes refratários
Azatioprina, Ciclofosfamida, Ciclosporina, Rituximab, Colchicina, Dapsona, entre outros.

28
Tratamento PTI crônica Esplenectomia Rituximab Vincristina
Célula B Tratamento Rituximab Vincristina Ciclofosfamida PTI crônica Macrófago plaqueta Esplenectomia Imunoglobulina Receptor IIb/IIIa – receptor de fibrinogênio Receptor da porção Fc da Imunoglobulina

29
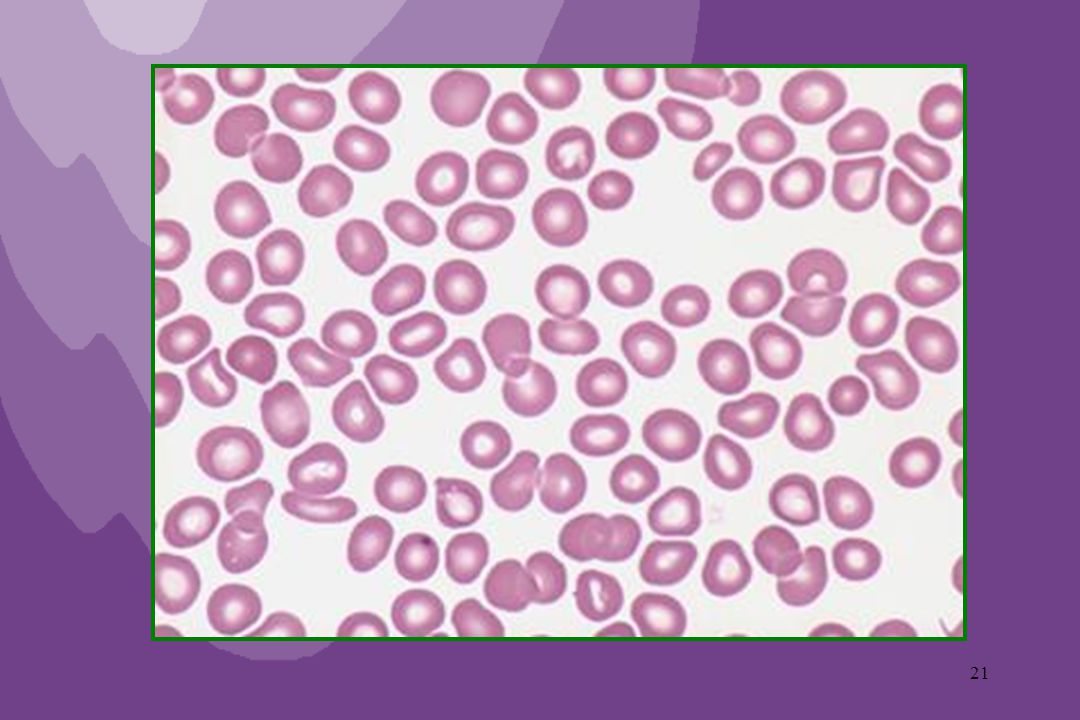
Microangiopatia Trombótica :
Púrpura trombocitopênica trombótica (D. de Moschcowitz) : Caracterizada pela oclusão difusa de arteríolas e capilares da microcirculação por microtrombos compostos basicamente de plaquetas, levando à isquemia dos tecidos. Ocorre principalmente em adultos sendo mais freqüentes em mulheres. Formas : Primária e Secundária (D. autoimunes – LES, AR). Síndrome hemolítico-urêmica : Idem, com predominância de quadro renal associado (IRenal Aguda) Ocorre principalmente em crianças. Formas : Primária e Secundária (Infecções).
 : Caracterizada pela oclusão difusa de arteríolas e capilares da microcirculação por microtrombos compostos basicamente de plaquetas, levando à isquemia dos tecidos. Ocorre principalmente em adultos sendo mais freqüentes em mulheres. Formas : Primária e Secundária (D. autoimunes – LES, AR). Síndrome hemolítico-urêmica : Idem, com predominância de quadro renal associado (IRenal Aguda) Ocorre principalmente em crianças. Formas : Primária e Secundária (Infecções).")
30
Microangiopatia Trombótica :
Sangue periférico : Histopatologia :

31
Púrpuras Vasculares Não trombocitopênicas
Resultantes de alterações relacionadas à integridade dos vasos Investigação laboratorial normal – hemograma e testes de hemostasia normais

32
Causas de Púrpura Vascular
Doenças do tecido conjuntivo: Escorbuto, uso de corticóide, amiloidose, S. de Marfan, S. de Ehlers-Danlos, senil Tóxica: viroses, meningococcemia, ricketsioses, púrpura fulminante Mecânica: esforço, leucostase, trombótica, embolia gordurosa Vasculite: colagenose, Henoch-Schonlein, cioglobulinemia Alteração da angiogênese: Telangectasia hemorrágica hereditária

33
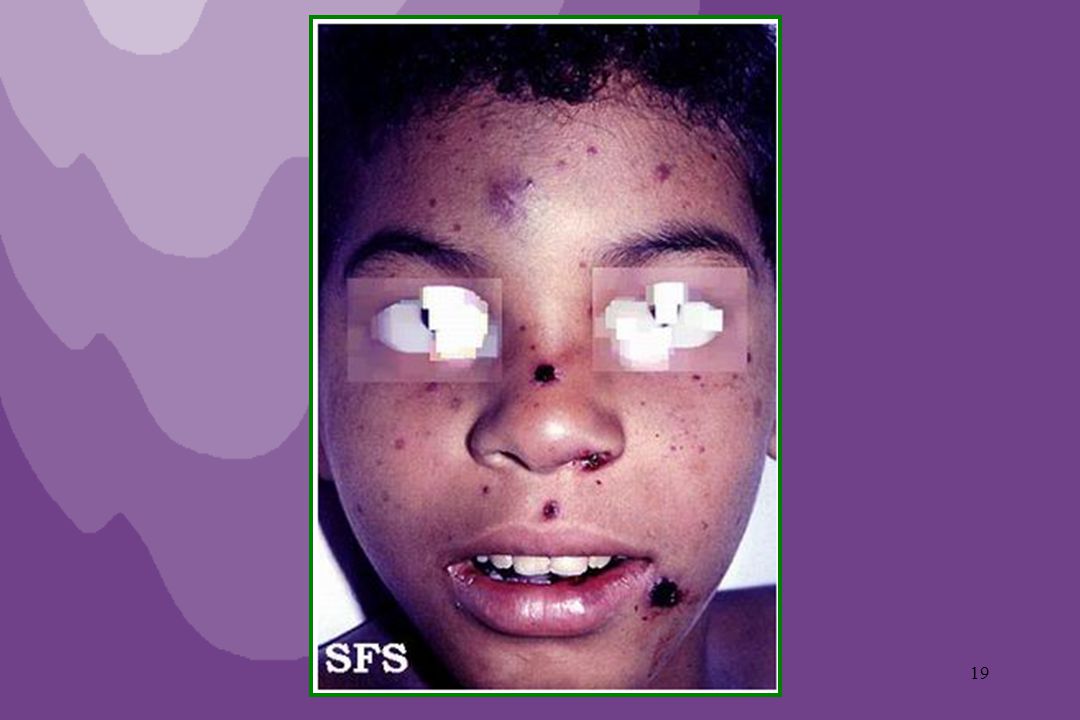
Púrpura de Henoch-Schonlein (Púrpura Anafilactoide)
Vasculite de hipersensibilidade, mediada por imunocomplexos. Desencadeada geralmente por infecções (estreptococcias, hepatite B, CMV, EBV), medicamentos (sulfa, alopurinol, penicilina, iodetos, cimetidina), produtos químicos (inseticidas, conservantes ou corantes de alimentos industrializados). Atinge principalmente crianças.
, medicamentos (sulfa, alopurinol, penicilina, iodetos, cimetidina), produtos químicos (inseticidas, conservantes ou corantes de alimentos industrializados). Atinge principalmente crianças.")
34
Púrpura de Henoch-Schonlein
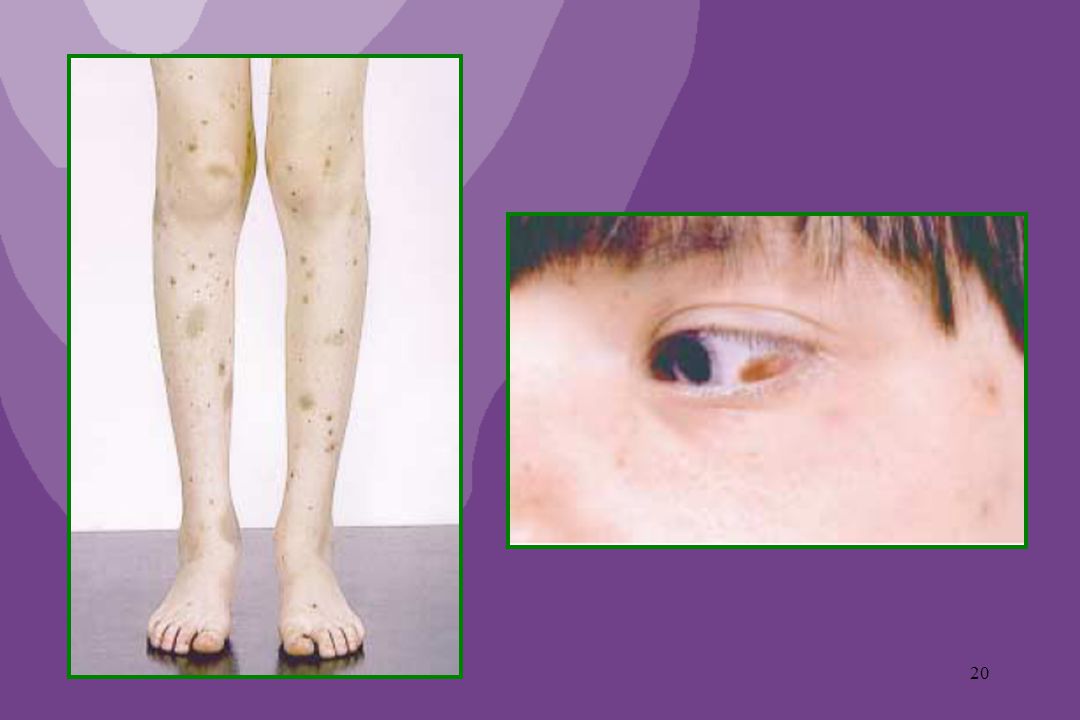
SINTOMAS: Lesões purpúricas elevadas, avermelhadas, por vezes com área de necrose isquêmica. Distribuição característica: ocorre geralmente em membros inferiores, bilateral, assimétrica, ascendendo progressivamente. Pode haver artralgia ou mesmo artrite, dor abdominal, cefaléia e hematúria.

35
Púrpura de Henoch-Schonlein
DIAGNÓSTICO Lesão purpúrica e sintomas associados (nem sempre presentes) Moderada redução do fator XIII, em correlação direta com a atividade da doença Eventualmente, biópsia é necessária para confirmação, principalmente nos casos persistentes (infiltrado inflamatório característico da vasculite leucocitoclástica) ou recorrentes.
 Moderada redução do fator XIII, em correlação direta com a atividade da doença. Eventualmente, biópsia é necessária para confirmação, principalmente nos casos persistentes (infiltrado inflamatório característico da vasculite leucocitoclástica) ou recorrentes.")
36
Púrpura de Henoch-Schonlein
Tratamento sintomático – corticosteróides aliviam os sintomas, mas não previne comprometimento renal Glomerulonefrite – complicação mais temível embora seja infreqüente e raramente comprometa a função a renal. Doença de curso benigno, na maioria das vezes resolve–se em 4 a 6 semanas Crises recorrentes – observar se houve nova exposição ao agente desencadeante ou pesquisar neoplasias ocultas.

37
Telangectasia Hemorrágica Hereditária (D. de Osler-Rendu-Weber)
Doença genética, de herança autossômica dominante Leva ao aparecimento de telangectasias observadas na região perioral, mucosa nasal, língua e leito ungueal Epistaxe recorrente e sangramento gastrintestinal são achados freqüentes, e costumam agravar-se com a idade Tratamento de suporte

Apresentações semelhantes





, e que pode levar a incapacitação funcional.>")





 Fase plaquetária (formação do trombo de plaquetas) Fase plasmática (formação do coágulo de fibrina)>")



 até o 5° mês de vida fetal l Destruição de hemácias envelhecidas (120.>")

>")


