Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Defeito Funcional Plaquetário Hereditário e Adquirido Reunião Clínica do CHSP
Nydia S Bacal 26, Setembro, 2007

2
Objetivo Morfologia plaquetária e hemostasia primária
Defeitos plaquetários hereditários e adquiridos Métodos laboratoriais no diagnóstico e classificação das disfunções plaquetárias

3
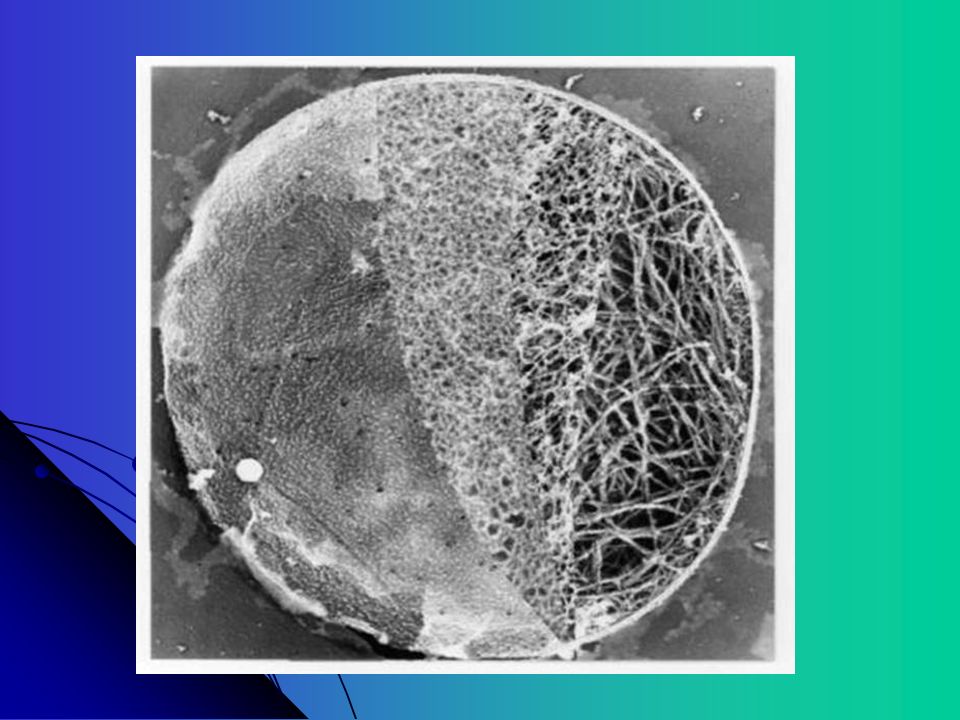
Plaqueta Célula discóide anuclear (3-5 microns).
Fragmentos do citoplasma de megacariócitos da medula óssea. Maturação 4-5 dias, meia vida 9-10 dias Membrana bilaminar que contem várias invaginações com um sistema canalicular aberto: Ligado intracelularmente a um sistema tubular denso, formando uma rede de interconexão através da célula (complexo membránico) Facilita a secreção dos grânulos
 Facilita a secreção dos grânulos.")
4
Célula não nucleada com meia-vida de 7 a 10 dias.
Pode reduzir a meia-vida para 3 a 4 dias indicando aumento de consumo, aumento do turnover plaquetário. (geneticamente? patológicamente?) Um espiral de microtubulos mantém a forma discóide da plaqueta. A membrana plaquetária estica como um guarda chuva em espiral e circula na corrente sanguínea. A estabilidade é conseguida por dois sistemas de canais inter-conectados no interior da plaqueta: a fina rede de ligação do canal –sistema tubular denso (amarelo) e os tubos tipo saco (azul) o qual também prove a conexão com o exterior. A plaqueta é altamente especializada e diferenciada. Além dos dois sistemas de canais apresenta outras organelas (mitocondrias, lisossomos, grânulos de estocagem de glicogênio, etc…).
 Um espiral de microtubulos mantém a forma discóide da. plaqueta. A membrana plaquetária estica como um guarda chuva em. espiral e circula na corrente sanguínea. A estabilidade é conseguida por dois sistemas de canais. inter-conectados no interior da plaqueta: a fina rede de ligação. do canal –sistema tubular denso (amarelo) e os tubos tipo saco. (azul) o qual também prove a conexão com o exterior. A plaqueta é altamente especializada e diferenciada. Além dos. dois sistemas de canais apresenta outras organelas. (mitocondrias, lisossomos, grânulos de estocagem de. glicogênio, etc…).")
6
Organelas da Plaqueta Mitocondria, Aparelho de Golgi, ribossomos, peroxisomos, lisossomos Dois tipos de Grânulos de armazenamento: Grânulos Alpha: Fator 4 Plaquetário , PDGF, fibrinogênio, fibronectina, inibidor I do ativador de plasminogênio (PAI I), Fatores V, VIII,e FvW Corpos Densos: histamina, epinefrina, serotonina, ADP, cálcio
, Fatores V, VIII,e FvW. Corpos Densos: histamina, epinefrina, serotonina, ADP, cálcio.")
7
membrana microtubulos Sistema tubular denso Sistema canalicular conectado a superfície Mitocondria Grânulos de glicogênio Grânulos Densos Grânulos α

8
Citoesqueleto plaquetário
Composto de filamentos de actina cruzados, ligados a superfície interna da dupla membrana lipídica Regula a forma da plaqueta em repouso Interage com os receptores transmembranicos Ativação plaquetária através da cascata de fosforilação proteica intracelular leva a sua contração e liberação do conteúdo das organelas plaquetárias

10
PLAQUETAS Sistema canicular aberto Sistema Tubular Denso Grânulo α
Microtubulos Grânulo Denso Filamentos Actínicos Estoque de glicogênio Miosina Mitocondria Degranulação – liberação do conteúdo dos grânulos α e densos Mudança de forma (discóide para esférica) e extensão de pseudópodes
 e extensão de. pseudópodes.")
11
Superfície plaquetária
Possui numerosos receptores G-proteicos ou receptores de adesão (integrinas): heterodímeros transmembranicos compostos de subunidades alpha e beta, responsáveis pela adesão e sinais de transdução São glicoproteinas designadas de I até IX (de forma decrescente conforme tamanho molecular); e de a e b conforme tipo de bandas obtidas por eletroforese
: heterodímeros transmembranicos compostos de subunidades alpha e beta, responsáveis pela adesão e sinais de transdução. São glicoproteinas designadas de I até IX (de forma decrescente conforme tamanho molecular); e de a e b conforme tipo de bandas obtidas por eletroforese.")
12
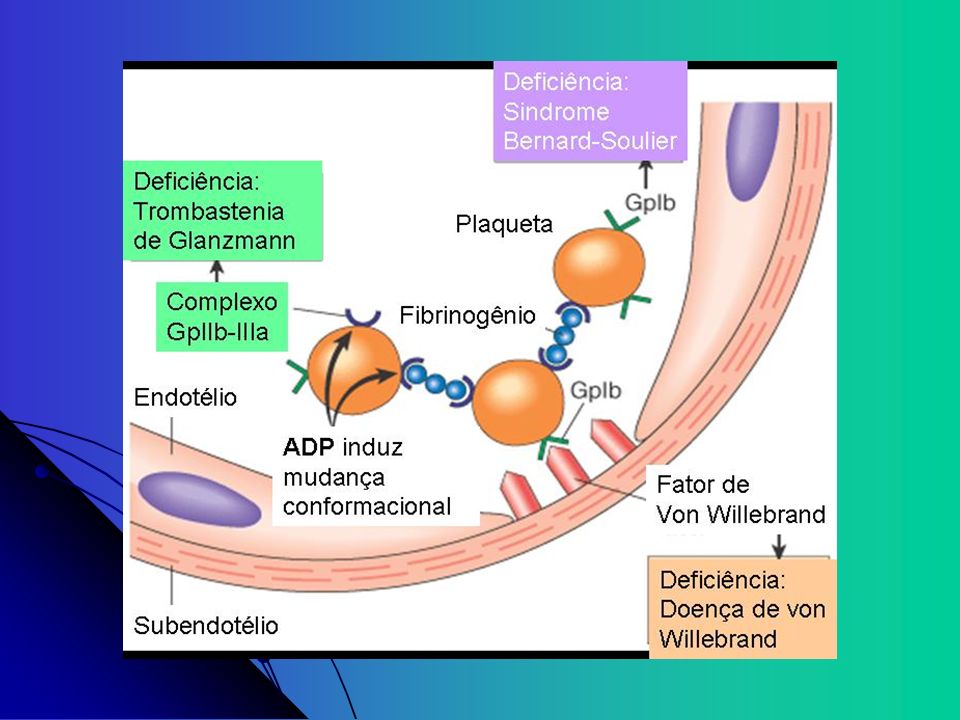
Glicoproteinas receptoras
GP Ib-V-IX: complexo de quatro produtos gênicos, serve como um receptor para FvW; adesão - (Bernard-Soulier) GP IIb-IIIa: mais abundante, reconhece os quatro receptores de adesão: fibrinogênio, fibronectina, vitronectina, e FvW; agregação - (Glanzmann) Outros: GP Ia, IIa; GP VI: receptores do colágeno
 GP IIb-IIIa: mais abundante, reconhece os quatro receptores de adesão: fibrinogênio, fibronectina, vitronectina, e FvW; agregação - (Glanzmann) Outros: GP Ia, IIa; GP VI: receptores do colágeno.")
13
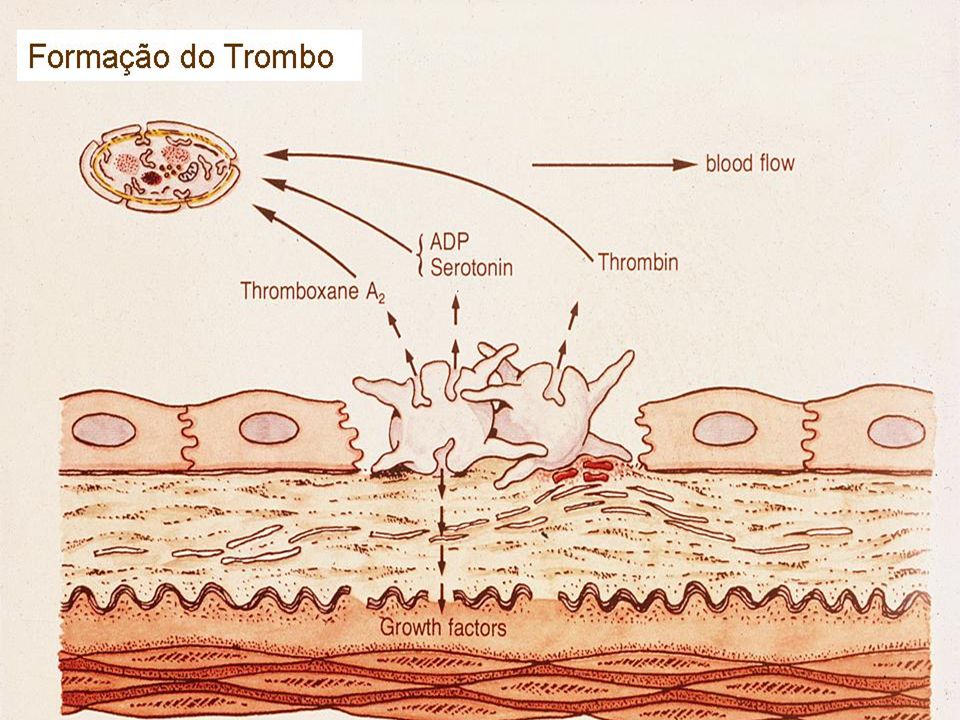
Hemostasia Primária Extremamente dinâmica, interação contínua entre vaso, plaqueta, e componentes do plasma Adesão, Ativação (Secreção), Agregação
, Agregação.")
14
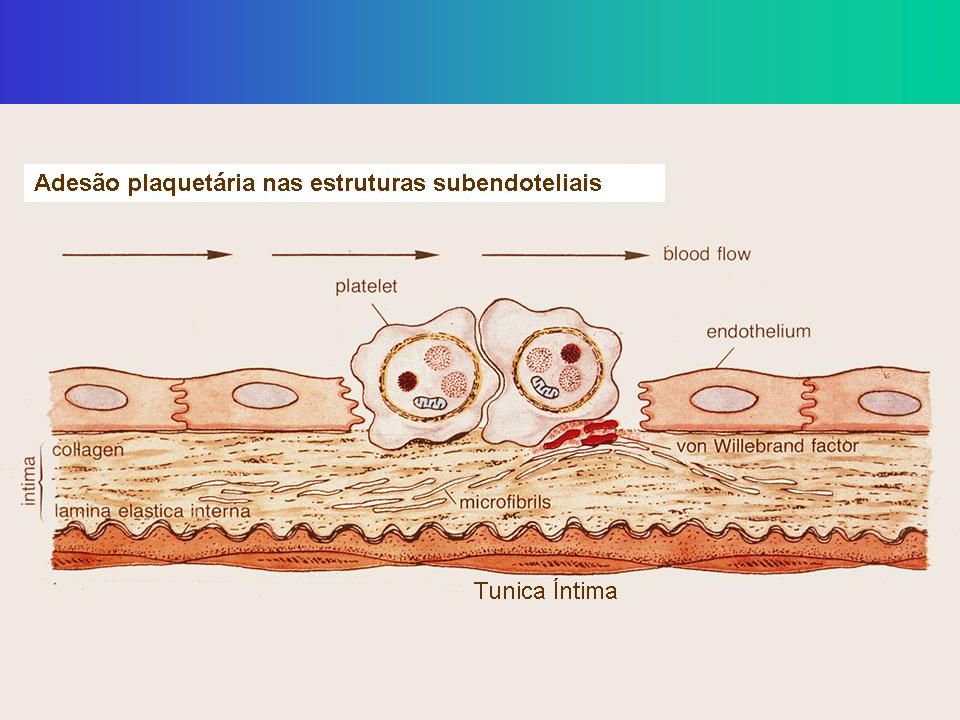
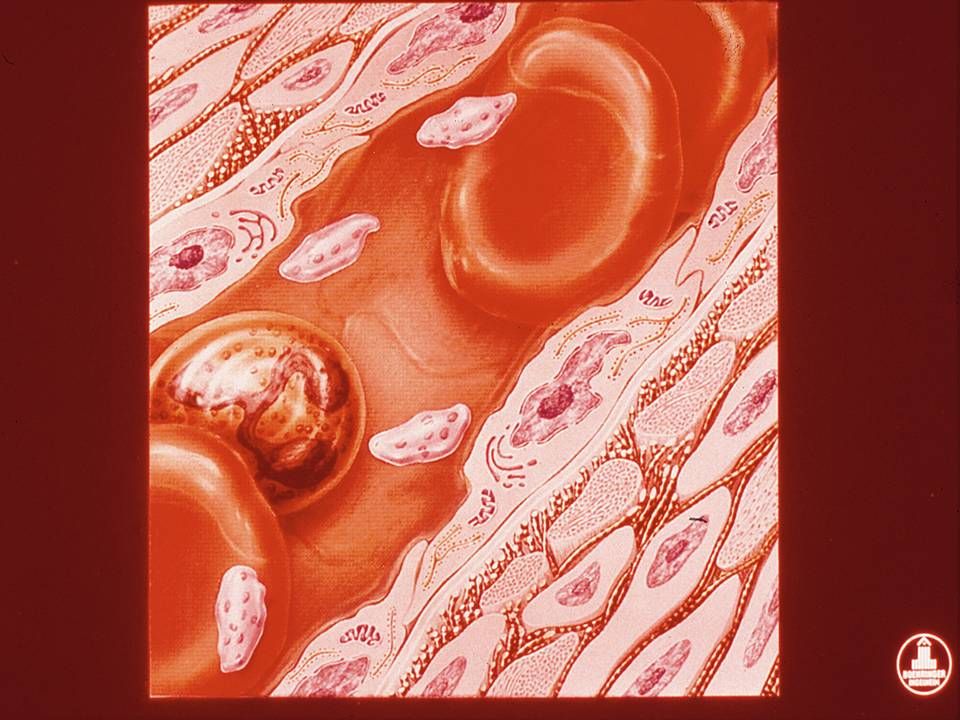
Adesão Rompimento vascular expõe os componentes pró-coagulantes do sub-endotélio (matrix extracelular, colágeno, proteoglicanos, e fibronectina); O FvW atua como uma ponte de adesão entre o complexo GP Ib-V-IX da plaqueta e o colágeno exposto; As plaquetas aderem-se a fibronectina das microfibrilas subendoteliais; A associação FvW-GPIb é a ligação mais forte para superar a força do fluxo sanguíneo.

16
Subendotélio

18
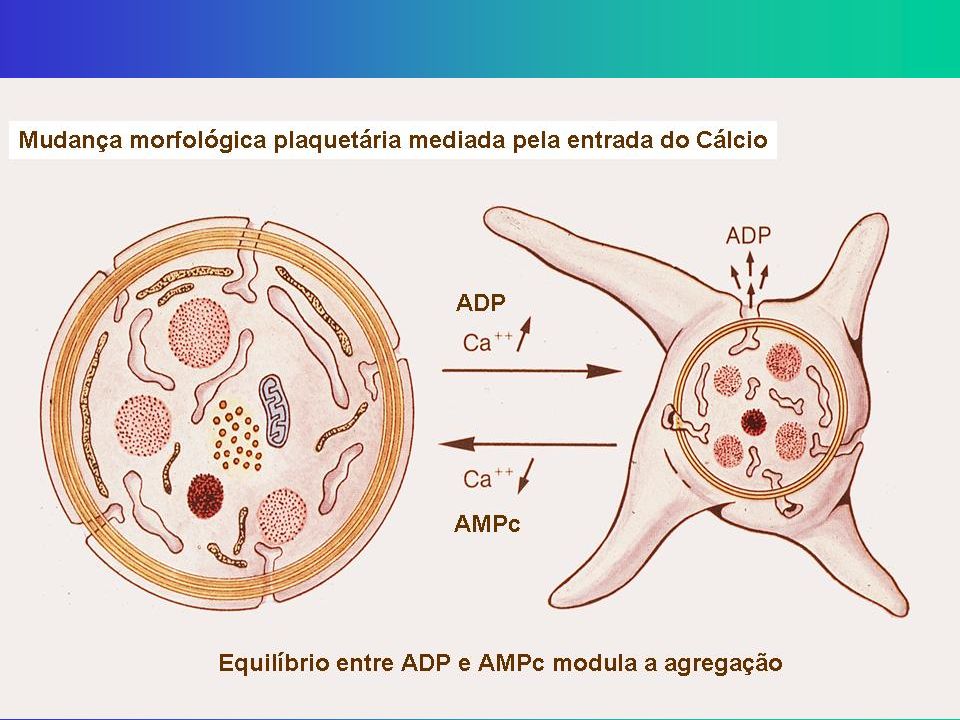
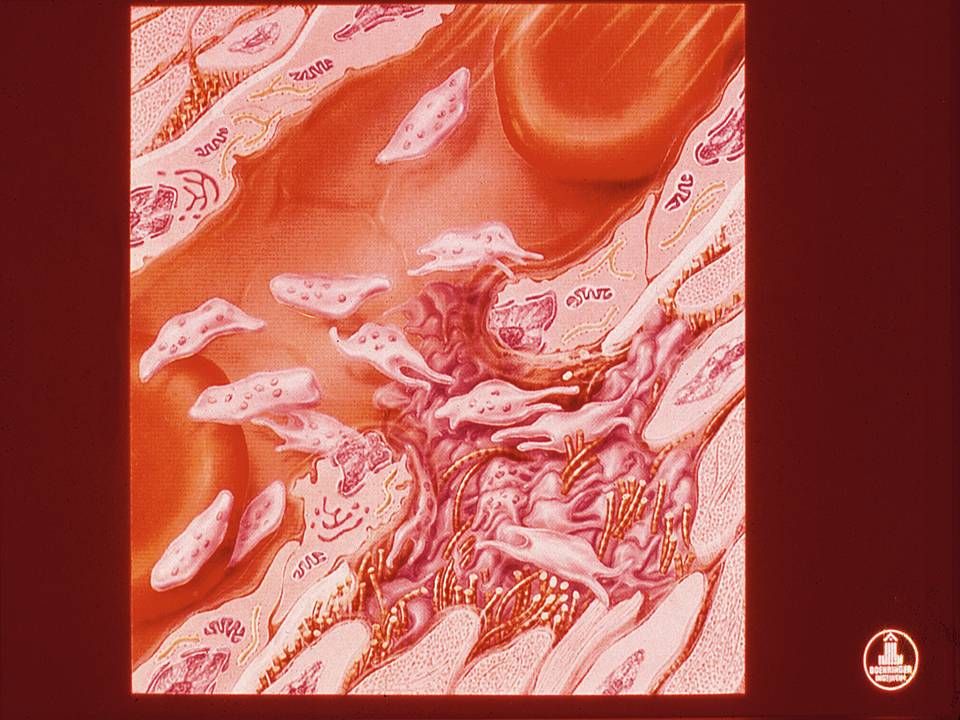
Secreção - Ativação Mudança da forma de discóide para esférica com extensão de pseudópodes promovida pela contração do citoesqueleto Conteúdo dos Granulos plaquetários são liberados através do sistema canalicular A contração do citoesqueleto ocorre pelo aumento do Ca iônico intraplaquetário, modulado pela redução do AMPc e aumento de ADP proveniente da via das prostaglandinas (TXA2) Também são expostos fosfolipídios na superfície da membrana plaquetária, que são receptores dos fatores da coagulação vitamina K dependentes (Fator II, IX, X)
 Também são expostos fosfolipídios na superfície da membrana plaquetária, que são receptores dos fatores da coagulação vitamina K dependentes (Fator II, IX, X)")
20
Contraste de Fase

21
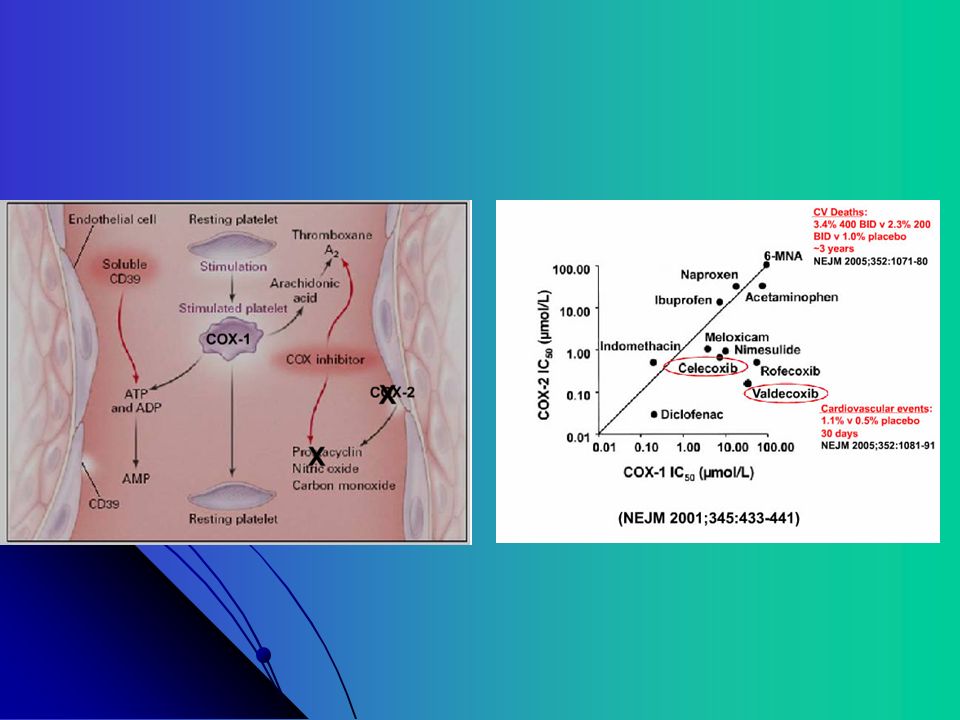
↓ Ca++ ↑ Via das Prostaglandinas (AAS) (Clopidogrel) Prostaglandinas
Ciclooxigenase (AAS) Prostaglandinas Tromboxane Sintetase (Clopidogrel) Prostaciclina Sintetase ↓ Ca++ ↑ Drogas Antiagregantes interferem nesses mecanismos Prostaciclina (PGI2): poderoso antiagregante plaquetário e vasodilatador Tromboxane A2: poderoso agregante plaquetário e vasoconstrictor
 Prostaglandinas. Tromboxane Sintetase. (Clopidogrel) Prostaciclina Sintetase. ↓ Ca++ ↑ Drogas Antiagregantes. interferem nesses. mecanismos. Prostaciclina (PGI2): poderoso antiagregante. plaquetário e vasodilatador. Tromboxane A2: poderoso agregante plaquetário e. vasoconstrictor.")
22
Agregação A contração da plaqueta provocada pela reação de liberação propicia: Mudança conformacional dos receptores IIbIIIa, permitindo a ligação do fibrinogênio Agregação plaquetária via fibrinogênio Reação auto-catalítica ativando outras plaquetas Formação do plug primário hemostático

29
PLAQUETOPATIAS CONGÊNITAS
Raras PORÉM... Diagnósticos errados - terapias erradas Corticóide, reposição de FvW , Esplenectomia, ou omissão de tratamento

30
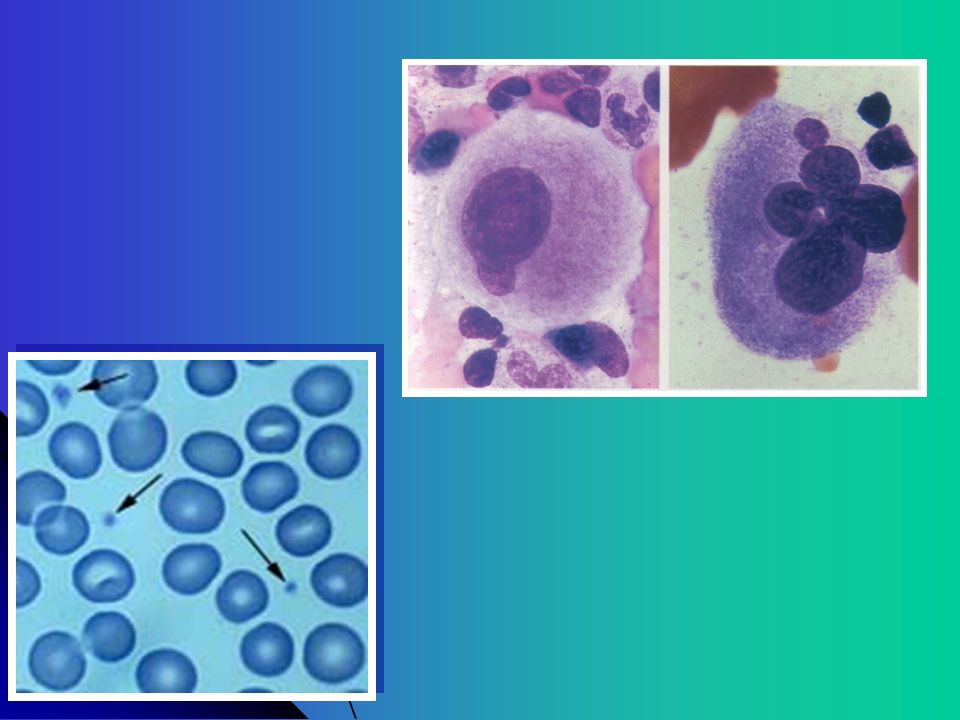
Quando Suspeitar ? História Familiar - Dois ou mais parentes afetados Sintomas associados - Defeitos Ósseos , Surdez, Catarata congênita, Eczema, Imunodeficiência, Alterações Neurológicas Sem resposta a tratamento para PTI Visualizar em Sangue Periférico – Inclusões em Neutrófilos, Plaquetas Gigantes, Alteração dos grânulos Sangramento desproporcional ao Nível Plaquetário Plaquetopenia há muitos anos Sangramento desde infância

31
EXAMES LABORATORIAIS NA AVALIAÇÃO PLAQUETÁRIA
Contagem Manual de Plaquetas Importante contadores automáticos não contam plaquetas com > 20 fL analisadores automaticos hematológicos com atc monoclonais Avaliar Morfologia Plaquetária Confirmar trombocitopenia Tamanho (macroplaquetas, microplaquetas) Grânulos plaquetários Tempo de Sangramento – Ivy Agregação Plaquetária - Técnica de Born PFA – 100 x TS Adesividade Plaquetária – Técnica de Salzman Impact – (Diamed) Citometria de Fluxo Proteínas de superfície plaquetária CD41 (GPIIb) e CD61 (GPIIIa), CD42b (GPIb), CD42a (GPIX), CD42d (GPV)
 Grânulos plaquetários. Tempo de Sangramento – Ivy. Agregação Plaquetária - Técnica de Born. PFA – 100 x TS. Adesividade Plaquetária – Técnica de Salzman. Impact – (Diamed) Citometria de Fluxo. Proteínas de superfície plaquetária CD41 (GPIIb) e CD61 (GPIIIa), CD42b (GPIb), CD42a (GPIX), CD42d (GPV)")
32
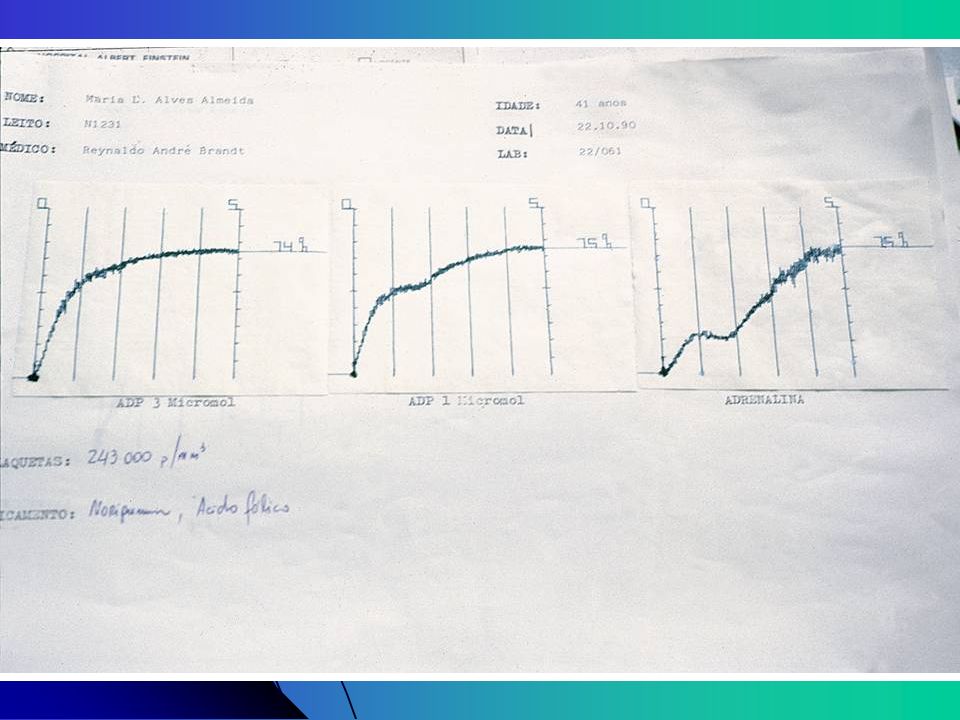
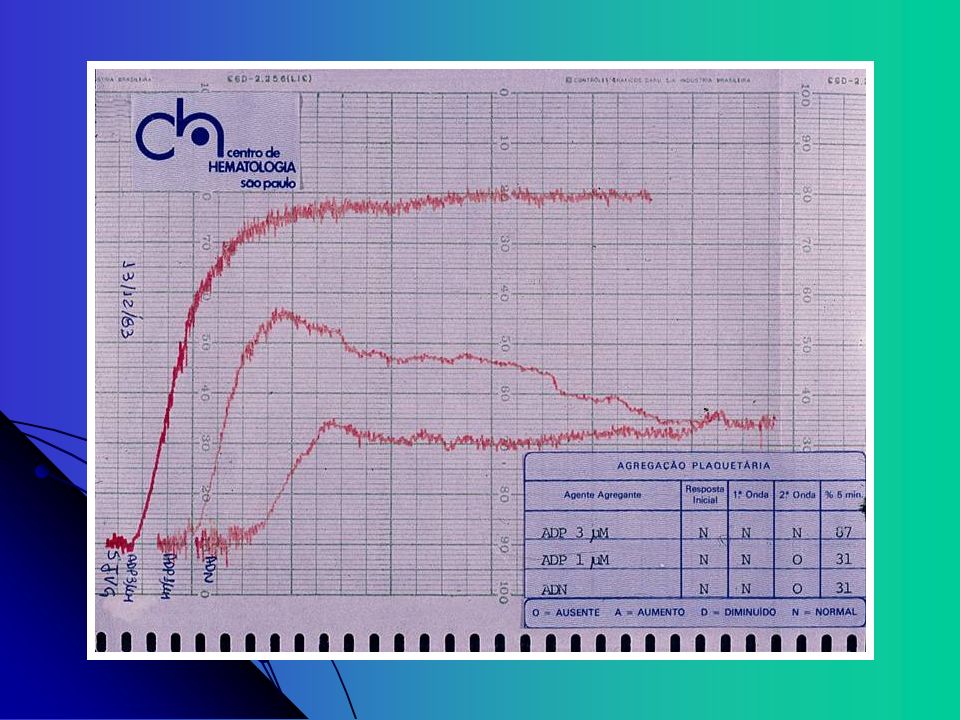
Estudo da Agregação Plaquetária Agentes agregantes
Concentrações diferentes de cada agente levam à formas diferentes de curvas ADP, ADN e Colágeno permitem a obtenção de curvas com duas ondas. Primeira onda: efeito direto do agente agregante Segunda onda: representa a reação de liberação plaquetária Ácido aracdônico Ristocetina (antibiótico): teste de aglutinação plaquetária dependente do FvW, pode ser realizada com plaquetas fixadas com formol Defeito na agregação induzida com Ristocetina é característico de Bernard-Soulier
: teste de aglutinação plaquetária dependente do FvW, pode ser realizada com plaquetas fixadas com formol. Defeito na agregação induzida com Ristocetina é característico de Bernard-Soulier.")
33
Estudo da Agregação Plaquetária
PRP é preparado em sangue total citratado por leve centrifugação (cerca de 500 rpm) Agonistas ou agentes Agregantes Aumento da transmi - tância da luz Registro cinético pelo Agregômetro
 Agonistas ou agentes. Agregantes. Aumento da transmi - tância da luz. Registro cinético pelo. Agregômetro.")
35
Problemas no estudo da agregação plaquetária
Numerosas variáveis afetam a agregação: Anticoagulante (citrato de sódio) Plaquetometia ajustada no PRP – a /mm3 Distribuição do tamanho plaquetário Variação circadiana (?) Relação com alimentação e atividade física Atividade física diminui a concentração do AMPc – plaquetas “mais sensíveis ao ADP”, pode ser visto na hiperagregaçãodos atletas Ht alterado - corrigir o anticoagulante na coleta Tempo não pode ultrapassar 4 horas da coleta Na agregação com adrenalina aguardar 30 minutos/60 minutos
 Plaquetometia ajustada no PRP – a /mm3. Distribuição do tamanho plaquetário. Variação circadiana ( ) Relação com alimentação e atividade física. Atividade física diminui a concentração do AMPc – plaquetas mais sensíveis ao ADP , pode ser visto na hiperagregaçãodos atletas. Ht alterado - corrigir o anticoagulante na coleta. Tempo não pode ultrapassar 4 horas da coleta. Na agregação com adrenalina aguardar 30 minutos/60 minutos.")
36
Bleeding Time - Ivy Função Vascular e Plaquetária Pressão: 40 mmHg
Local: face anterior do antebraço Lanceta 1cm / 1mm de profundidade Papel de filtro a cada 30seg., evitando tocar as bordas do corte Duas incisões: divergência acima de 1 min devemos investigar a causa Valores normais: 7 a 9 minutos

37
PFA-100® Platelet Function Analyzer
Tempo de Fechamento do Orifício (obstrução) Cartucho com 0,8mL sg citratado Introdução do sg em alta velocidade através de um capilar que encontra uma membrana recoberta com colágeno / epinefrina ou colágeno / ADP que provoca adesão, ativação e agregação plaquetária Obstrui um orifício de 150 µm cessa o fluxo sg. Tempo de fechamento máx: 300 seg. Afetado pelo número de plaquetas e Hematócrito, Sensível ao Fator von Willebrand Não é afetado pelo fibrinogênio, Não apresenta correlação clínica em tromboses. C.P.M.Hayward et al – PFA-100 in the evauation of platelet disorders and platelet function- J. Thromb Haemost 2006; 4:312-9.
 Cartucho com 0,8mL sg citratado. Introdução do sg em alta velocidade. através de um capilar que encontra. uma membrana recoberta com. colágeno / epinefrina ou. colágeno / ADP que provoca adesão, ativação e agregação plaquetária. Obstrui um orifício de 150 µm. cessa o fluxo sg. Tempo de fechamento máx: 300 seg. Afetado pelo número de plaquetas e Hematócrito, Sensível ao Fator von Willebrand. Não é afetado pelo fibrinogênio, Não apresenta correlação clínica em tromboses. C.P.M.Hayward et al – PFA-100 in the evauation of platelet disorders and platelet function- J. Thromb Haemost 2006; 4:")
38
TS x PFA-100 Sensibilidade:
Baixa sensibilidade na triagem de pacientes com sangramento mucocutâneo Sensibilidade: TS 36% (D. do Pool de Armazenamento) PFA % (D. Von Willebrand) Combinação 48% Boa especificidade Especificidade: TS 91% PFA-100 (Epinefrina) 84% PFA-100 (ADP) 87% Thromb Haemost 2004:04:892 Thromb Haemost 2003:90:483
 PFA % (D. Von Willebrand) Combinação 48% Boa especificidade. Especificidade: TS 91% PFA-100 (Epinefrina) 84% PFA-100 (ADP) 87% Thromb Haemost 2004:04:892. Thromb Haemost 2003:90:483.")
39
Jenninngs L, White MM, Jacoski MV, Krihnat R-Journal of Thrombosis and Haemostasis, 2003

40
IMPACT IMPACT-R IMPACT (immediate microscopic platelet
Mede a função plaquetária sob pressão de atrito Sensível a função plaquetária,FvW,Ht e plaquetas IMPACT (immediate microscopic platelet adhesion cone and plate technology) DiaMed Based upon cone and platelet viscometer developed by Varon A 130ul of whole blood (citrated) is placed in a polystyrene plate Shear rate of 1800/s is applied for 2 min Platelets adhere and aggregate on the plate surface Washing and staining The plate is placed under a microscope connected to an image analysis system and a computer Successive images of plate are taken and the percentage of the well covered by the stained objects and the average size of these objects are quantified IMPACT IMPACT-R Kenet,G.A. et al - Br.J.Haematol :
 DiaMed. Based upon cone and platelet viscometer developed by Varon. A 130ul of whole blood (citrated) is placed in a polystyrene plate. Shear rate of 1800/s is applied for 2 min. Platelets adhere and aggregate on the plate surface. Washing and staining. The plate is placed under a microscope. connected to an image analysis system and a computer. Successive images of plate are taken and the percentage of the well covered by the stained objects and the average size of these objects are quantified. IMPACT IMPACT-R. Kenet,G.A. et al - Br.J.Haematol :")
41
TEG Diamandis M et al.- J Thromb Haemost May;4(5):
:")
42
O anticorpo CD42b é diretamente ligado a glicoproteina plaquetária GPIb está
ausente ou presente em baixa expressão em pacientes com Síndrome de Bernard-Soulier . A glicoproteina plaquetária GPIb atua como receptor para o Fator Von Willebrand e apresenta uma alta afinidade ao receptor de trombina. Raramente utilizamos o CD42a(GPIX) e o CD42d(GPV) . Na Trombastenia de Glanzmann há diminuição ou ausência do GP IIb (CD41) e o GP IIIa (CD61)
 e o CD42d(GPV) . Na Trombastenia de Glanzmann há diminuição ou ausência do GP IIb (CD41) e o. GP IIIa (CD61)")
43
Trombastenia de Glanzmann
Eduard Glanzmann ( ), pediatra suiço Relata um caso de doença com sangramento inicial imediatamente após o nascimento W. E. Glanzmann:Hereditäre hämorrhägische Thrombasthenie. Ein Beitrag zur Pathologie der Blutplättchen. Jahrbuch für Kinderheilkunde, 1918; 88: 1-42,
, pediatra suiço. Relata um caso de doença com sangramento inicial imediatamente após o nascimento. W. E. Glanzmann:Hereditäre hämorrhägische Thrombasthenie. Ein Beitrag zur Pathologie der Blutplättchen. Jahrbuch für Kinderheilkunde, 1918; 88: 1-42,")
44
Trombastenia de Glanzmann – Quadro Clínico
Menorragia Equimoses Epistaxe Gengivorragia Hemorragia TGI Hematúria Hemartrose SNC Hematoma Visceral 98% 86% 73% 55% 12% 6% 3% 2% 1% George JN et al., Glanzmann’s thrombastenia: The spectrum of clinical disease. Blood. 1990, 75:1383

45
Diagnóstico- Trombastenia de Glanzmann
Plaquetometria e morfologia são normais Tempo de Sangramento prolongado Redução ou ausência da agregação plaquetária em resposta a múltiplos agonistas: ADP,AND, trombina, ou colágeno (exceção Ristocetina) Citometria de Fluxo: diminuição da expressão do Atc Mono CD41 (GPIIb) e CD61 (GPIIIa)
 Citometria de Fluxo: diminuição da expressão do Atc Mono CD41 (GPIIb) e CD61 (GPIIIa)")
46
Trombastenia – Doença de Glanzmann - Naegeli
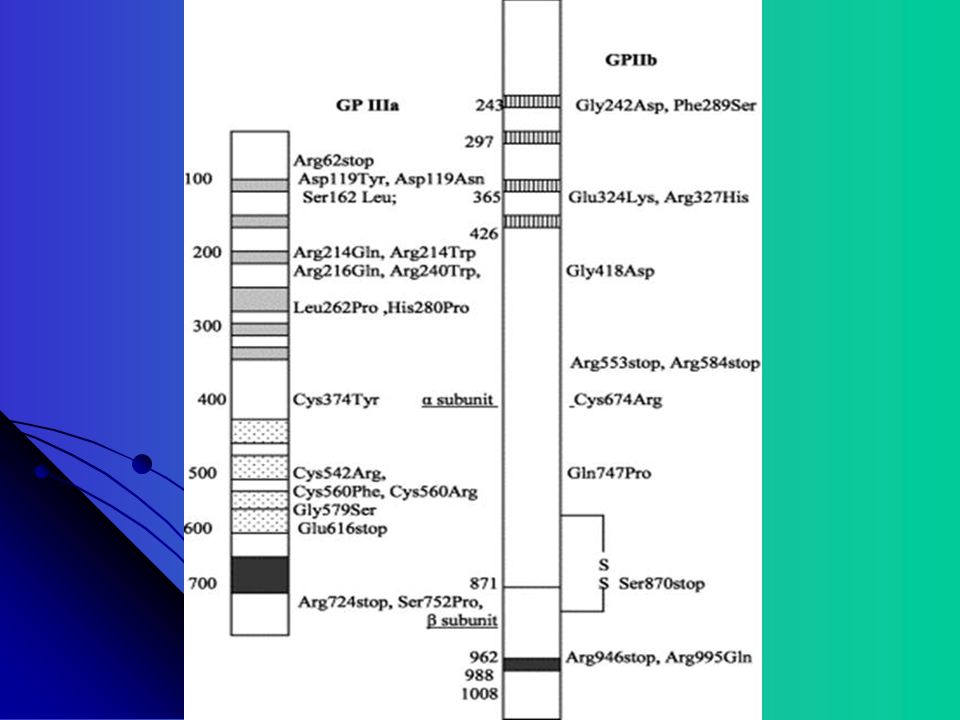
Receptor da superfície plaquetária mais abundante IIbIIIa (80,000 por plaqueta) Complexo IIbIIIa é um heterodímero dependente do Ca++ Genes para ambas subunidades são encontrados no Cromossomo 17 Doença Autossômica Recessiva causada por mutações (substituição, inserção, deleção) em genes codificados para IIb ou IIIa, resultando em anormalidades qualitativas ou quantitativas das proteinas
 Complexo IIbIIIa é um heterodímero dependente do Ca++ Genes para ambas subunidades são encontrados no Cromossomo 17. Doença Autossômica Recessiva causada por mutações (substituição, inserção, deleção) em genes codificados para IIb ou IIIa, resultando em anormalidades qualitativas ou quantitativas das proteinas.")
48
O defeito fundamental do paciente com trombastenia é a inabilidade da plaqueta em se agregar
Outro problema: a plaqueta não se adere normalmente na matrix do subendotélio (devido a perda da interação da fibronectina/FvW e IIbIIIa ) Também, o fibrinogênio dos grânulos alpha está diminuído ou ausente
 Também, o fibrinogênio dos grânulos alpha está diminuído ou ausente.")
49
Pacientes com Glanzmann são classificados em três grupos baseados na expressão do complexo:
Tipo I menos de 5% GPIIbIIIa, ausência de fibrinogênio no grânulo alpha Frequentemen resultado da mutação do gene IIb Tipo II <20%, fibrinogênio presente Tipo III >50%, trombastenia “variante”, doença qualitativa

50
Síndrome de Bernard-Soulier
Primeiro descrito em 1948 por Jean Bernard and Jean-Pierre Soulier; França Bernard J, Soulier JP: Sur une nouvelle variete de dystrophie thrombocytaire hemarroagipare congenitale. Sem Hop Paris 24:3217, 1948 Autossômica Recessiva; caracterizada por trombocitopenia moderada a severa , plaquetas gigantes, e sangramento espontâneo Caracterizada pela deficiência ou disfunção do complexo GP Ib-V-IX que perdem a capacidade de aderir ao FvW

51
Síndrome Bernard-Soulier
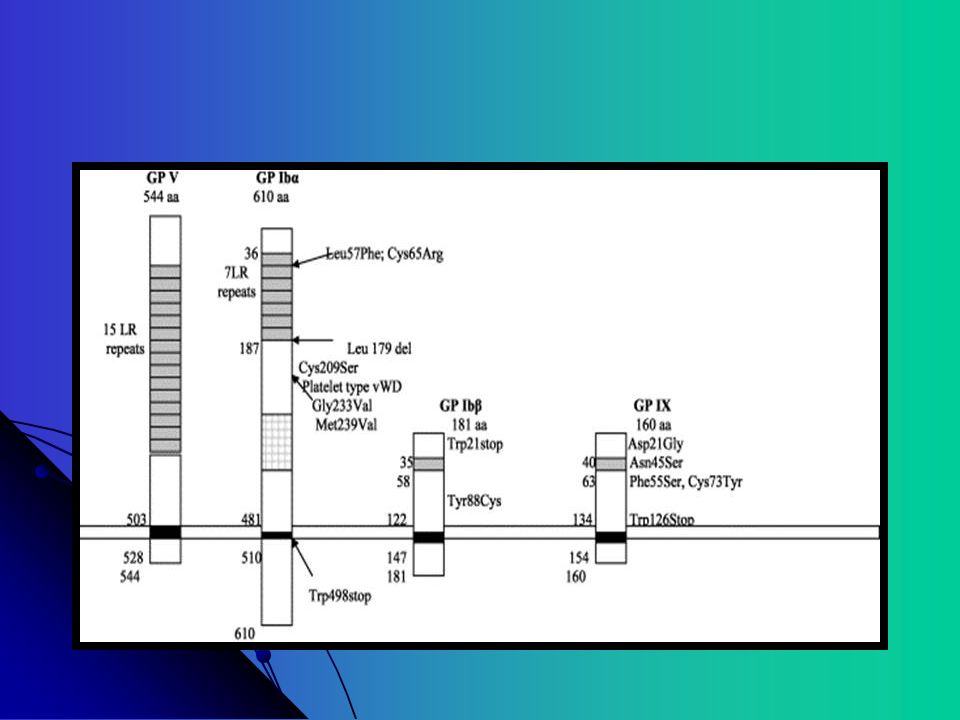
A plaqueta contem cerca de copias do GP Ib-V-IX GP 1b é um heterodímero com uma subunidade alpha e uma subunidade beta O gene para GP Ib alpha está localizado no cromossomo 17; GP Ib beta: cromossomo 22; GPIX e V: cromossomo 3 Muitas mutações são incompletas ou parciais resultando em parada precoce da codificação Muitas mutações envolvem a expressão do GP Ib, raramente a mutação do GP IX e não foi descrita nenhuma mutação no GP V

52
Síndrome de Bernard Soulier – Quadro Clínico
Epistaxe Equimoses Menorragia Gengivorragia Hemorragia TGI Pós-Trauma Hematúria SNC Sangramento Retina 70% 58% 44% 42% 22% 13% 7% 4% 2% Coller BS et al., Hereditary Qualitative Platelet Disorders. In: Williams Hematology, 2005, 7th Edition, McGraw-Hill, p

53
Diagnóstico- S. Bernard Soulier
T. S. prolongado (> 20 min), Trombocitopenia (plaqueta <20.000/mm3), Plaquetas gigantes (diâmetro médio >3.5 microns)
, Trombocitopenia (plaqueta <20.000/mm3), Plaquetas gigantes (diâmetro médio >3.5 microns)")
54
Diagnóstico – S. Bernard Soulier
Agregação plaquetária normal com todos os agonistas, exceção com a Ristocetina (oposto da Trombastenia) Citometria de Fluxo: diminuição da expressão antigênica para a GPIb – CD42b GPIX – CD42a GPV – CD42d
 Citometria de Fluxo: diminuição da expressão antigênica para a. GPIb – CD42b. GPIX – CD42a. GPV – CD42d.")
55
Síndrome de Bernard Soulier – Sindromes Correlatas
Macrotrombocitopenia do Mediterrâneo Pacientes da região do mediterrâneo com disfunção hemorrágica, macrotrombocitopenia Heterozigose para variante de Bolzano da Síndrome de Bernard-Soulier Síndrome de DiGeorge Síndrome genética em que há deleção do locus 22q11 CATCH 22 anormalidades faciais, cardíacas, defeitos de células T, hipoparatireodismo Deleção gene da Gp Ib – no locus 22q11.2 Muitos pacientes tem macrotrombocitopenia Sem diátese hemorrágica na maioria dos casos Doença de Von Willebrand Tipo Plaquetário Mutação na Gp Iba no sítio de ligação com o FvW Leva à ganho de função da Gp Iba consumo de plaquetas e FvW Macrotrombocitopenia e fenótipo semelhante à FvW tipo 2b Diagnóstico diferencial utilizando agregação com plaquetas do paciente ressuspensas em plasma normal.

56
Síndrome do Gene MYH9 Grupo de doenças de herança autossômica dominante que tem mutação do gene MYH9: - Anomalia de May-Hegglin (AMH) - Síndrome de Sebastian (SBS) - Síndrome de Epstein (EPTS) - Síndrome de Fechtner (FTNS) Tríade: Plaquetopenia, Plaquetas Gigantes e Inclusão em Neutrófilos tipo corpúsculo de Döhle. Características sindrômicas associadas: surdez, catarata, glomerulopatia renal. Distúrbios hemorrágico de leve intensidade ou ausente.
 - Síndrome de Sebastian (SBS) - Síndrome de Epstein (EPTS) - Síndrome de Fechtner (FTNS) Tríade: Plaquetopenia, Plaquetas Gigantes e Inclusão em Neutrófilos tipo corpúsculo de Döhle. Características sindrômicas associadas: surdez, catarata, glomerulopatia renal. Distúrbios hemorrágico de leve intensidade ou ausente.")
57
Doenças do Gene MYH9 Gene MYH9 localiza-se no cromossomo 22q11
Codifica proteína da cadeia pesada da miosina não muscular IIA (NMMHC-IIA) presente em leucócitos e plaquetas Papel na função do citoesqueleto fagocitose e formação plaquetária Descritas 20 diferentes mutações em 65 famílias
 presente em leucócitos e plaquetas. Papel na função do citoesqueleto fagocitose e formação plaquetária. Descritas 20 diferentes mutações em 65 famílias.")
58
Doenças do Gene MYH9 - Quadro Laboratorial
Plaquetopenia – /mm3 Presença de macrotrombócitos - maiores plaquetas descritas, cerca de 1/3 tem entre fL Histograma de Plaquetas Normais Plaquetas MYH9 Mecanismo envolvido – trombopoese ineficaz Disfunção plaquetária incapacidade de alterar sua forma por disfunção grave do citoesqueleto.

59
Doenças do Gene MYH9 Plaqueta Gigante e Corpúsculo tipo – Döhle

60
Corpúsculos tipo Döhle e IHQ para NMMHC-IIA
Doenças do Gene MYH9 Corpúsculos tipo Döhle e IHQ para NMMHC-IIA

61
Doença do Pool de Armazenamento
Classificada pelo tipo de deficiência: Agregação plaquetária semelhante à AAS Deficiência de corpos densos, Deficiência de grânulos alpha: Síndrome da Plaqueta Cinzenta, Deficiências mistas GP V – Plaquetopatia de Quebec

62
Deficiência de Corpos Densos
Corpos densos estão diminuídos (ADP, ATP, Cálcio, pirofosfato, 5HT) Contem 3-6 corpos densos de 300 micron
 Contem 3-6 corpos densos de 300 micron.")
63
Doenças hereditárias:
Síndrome de Hermansky-Pudlak Síndrome Wiskott-Aldrich Síndrome de Chediak-Higashi Síndrome da ausência de rádio e trombocitopenia (TAR)
")
64
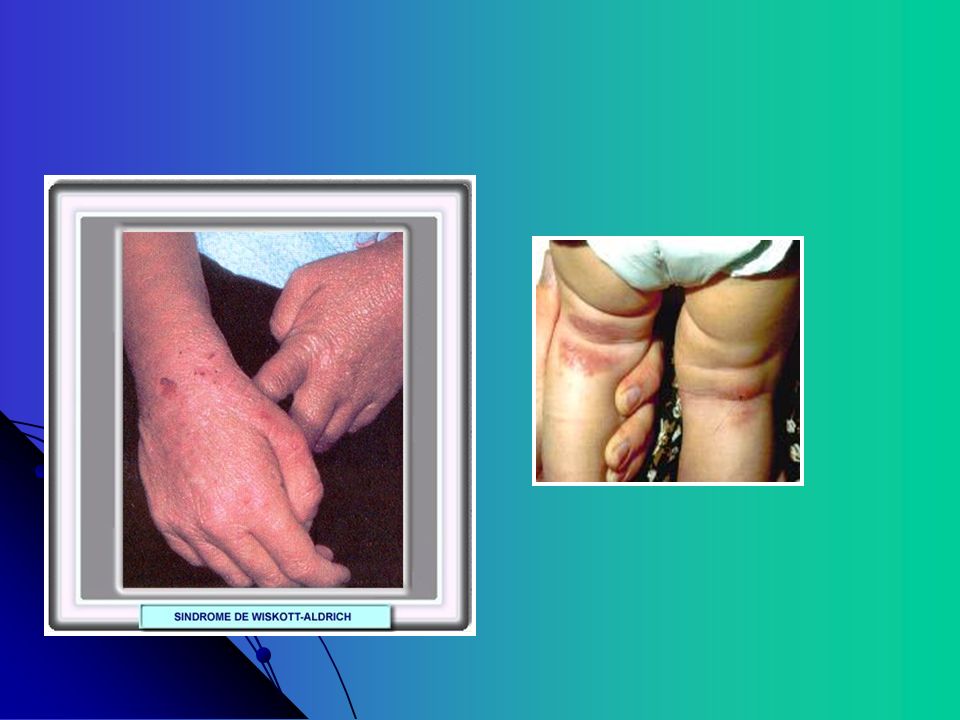
Wiskott-Aldrich Ligada X, defeito genético da proteina responsável pela actina na formação do citoesqueleto, proveniente de células hematopoéticas Caracterizada por trombocitopenia com defeito no pool de armazenamento plaquetário, eczema, e infecções recorrentes Sobrevida mediana anos. Fenômenos auto-imunes - doença inflamatória intestinal, artrite. Doenças linfoproliferativas Tratamento - Transplante de Medula óssea - Corticoesteróides

66
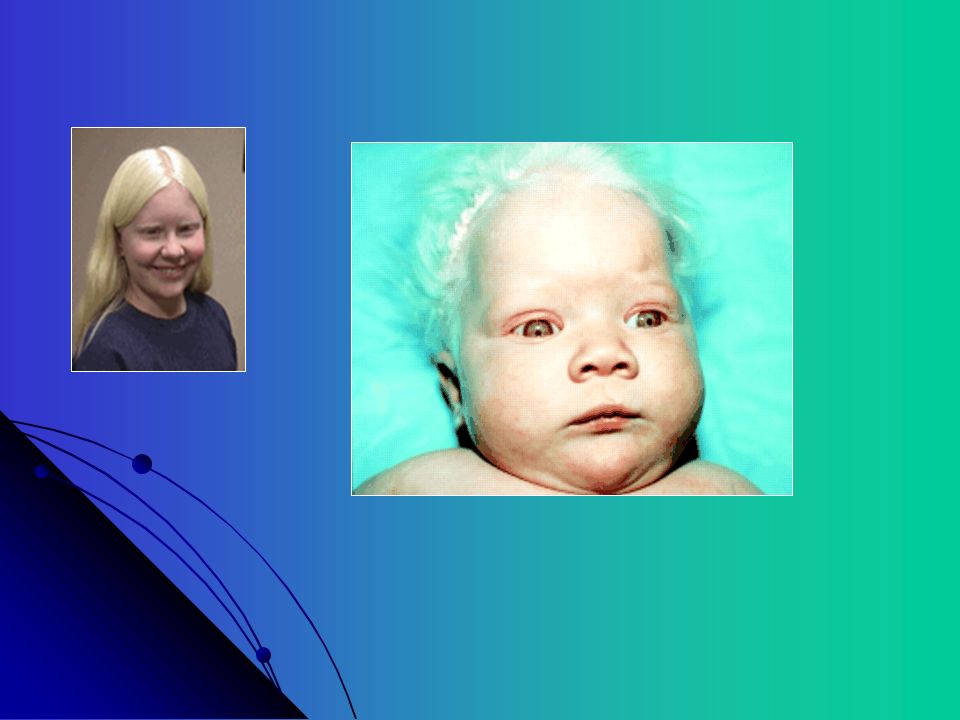
Hermansky-Pudlak Descrita em 1959 por Hermansky e Pudlak
Autossômica Recessiva, albinismo oculocutâneo, depósito tipo ceróide em lisossomos do SRE e medula óssea Associada com Fibrose pulmonar e infecções recorrentes Deficiência quantitativa dos grânulos densos com diátese hemorrágica leve a moderada Alta prevalência em Porto Rico

68
Depósito tipo ceróide

69
Chediak-Higashi Descrito por Beguez Cesar em 1943, Steinbrinck em 1948, Chédiak em 1952, e Higashi em 1954 Autossômica Recessiva; formação microtubular anormal e grânulos lisossomais gigantes presentes em fagócitos e melanócitos Ausência de degranulação / quimiotaxia = infecções bacterianas recorrentes Albinismo oculocutâneo parcial Diminuição ou ausência de grânulos nos corpos densos

71
Inclusões gigantes em neutrófilos

72
Trombocitopênia com ausência de Rádio (TAR)
Descrita em 1951 Autossômica Recessiva, caracterizada por ausência de radio, trombocitopenia (com defeito no pool de armazenamento), e outras anormalidades no esqueleto,no sistema GI, sistema cardiovascular Hemorragia é a maior causa de mortalidade Prognóstico melhor se sobreviver por 2 anos Etiologia não esclarecida
, e outras anormalidades no esqueleto,no sistema GI, sistema cardiovascular. Hemorragia é a maior causa de mortalidade. Prognóstico melhor se sobreviver por 2 anos. Etiologia não esclarecida.")
74
Trombocitopenia com Ausência de Rádio
Greenhalg et al. Thrombocytopenia-absent radius syndrome: a clinical genetic study, J Med Genet 2002;39:876–881

75
hematopoiese e morfogênese
Trombocitopenia com Sinostose Radio-Ulnar Mutação no gene HOXA11 – hematopoiese e morfogênese Thompson AA et al., Congenital thrombocytopenia and radio-ulnar synostosis: a new familial syndrome. Br J Haematol Jun;113(4):866-70
:")
76
Diagnóstico Estudo da agregação plaquetária está diminuida com baixa concentração de colágeno ADP e epinefrina mostram diminuição na resposta da segunda onda Agregação normal com Ristocetina Microscopia Eletrônica: perda de corpos densos

77
Síndrome da Plaqueta Cinzenta Deficiência de Granulos Alpha
Descrita por Raccuglia em 1971 Caracterizada pela redução dos grânulos alpha nas plaquetas. (plaquetas normais contem em média 50 grânulos alpha). Apresentam sangramento cutâneo mucoso leve a moderado
. Apresentam sangramento cutâneo mucoso leve a moderado.")
78
Diagnóstico Tempo de Sangramento prolongado, trombocitopenia moderada
Plaquetas grandes, acinzentadas e agranulares (ausência de cromômero) Estudo da Agregação: diminuição ou ausência de resposta ao colágeno
 Estudo da Agregação: diminuição ou ausência de resposta ao colágeno.")
79
PLAQUETOPATIAS ADQUIRIDAS
Drogas Uremia Cirrose Mieloma Múltiplo Paraproteina interfere na interação plaquetária Doença Mieloproliferativa(P.V. e T.E.) Grandes multímeros FvW adsorvem na plaqueta Bypass Cardiopulmonar Plaquetas em Banco de Sangue Dispende GP Ibα, produz ácido lático, ↓pH, hiporeatividade plaquetária, ↑ ATP, ativação com liberação do conteúdo granular, ↑ formação de trombina
 Grandes multímeros FvW adsorvem na plaqueta. Bypass Cardiopulmonar. Plaquetas em Banco de Sangue. Dispende GP Ibα, produz ácido lático, ↓pH, hiporeatividade plaquetária, ↑ ATP, ativação com liberação do conteúdo granular, ↑ formação de trombina.")
80
DIAGNÓSTICO Sem história em parentes de primeiro grau
dois locais/ um local com necessidade transfusional/ um local três vezes Exame de esfregaço sanguíneo TS / Teste com PFA Descartar Doença de Von Willebrand Agregação plaquetária Citometria de Fluxo Microscopia eletrônica para plaqueta ISTH SSC Subcommittee on VWD. J Thromb Haemost 2005:3:

81
1450 agregações plaquetárias realizadas
Trabalho apresentado na SBPC

82
Estudos Farmacogênicos
ISLH Miami – Aula Dra K.Kottke-Marchant

83
Bases Moleculares de Resistência a Agentes
Anti- Plaquetários Aspirina e Clopidogrel Usadas na prevenção de complicações ateroscleróticas Efeito terapêutico é heterogêneo Alguns pacientes não mostram inibição plaquetária esperada ou benefício com a terapia Variação: 5 a 60% Bases genômica ou proteômica não definida

84
Fontes Glassy, Eric ed. Color Atlas of Hematology: An Illustrated Field Guide Based on Proficiency Testing. (Northfield, Illinois: College of American Pathologists, 1998) Ramasamy I. Inherited bleeding disorders: disorders of platelet adhesion and aggregation. Crit Rev Oncol Hematol Jan;49(1):1-35. Review. Janeway CM, Rivard GE, Tracy PB, Mann KG. Factor V Quebec revisited. Blood May 1;87(9): Robbins. Pathologic Basis of Disease. 7th ed (2004) Hoffman, et al. Hematology: Basic Principles and Practice, 3r ed (2000) Aula Dra Kandice Kottke Marchant – ISLH Miami Wintrobe’s - Clinical Hematology 10 th ed. Lea &Febiger Bennett S J. – Inherited Disorders of Platelet Function - ASH/2005
 Ramasamy I. Inherited bleeding disorders: disorders of platelet adhesion and aggregation. Crit Rev Oncol Hematol Jan;49(1):1-35. Review. Janeway CM, Rivard GE, Tracy PB, Mann KG. Factor V Quebec revisited. Blood May 1;87(9): Robbins. Pathologic Basis of Disease. 7th ed (2004) Hoffman, et al. Hematology: Basic Principles and Practice, 3r ed (2000) Aula Dra Kandice Kottke Marchant – ISLH Miami Wintrobe’s - Clinical Hematology 10 th ed. Lea &Febiger. Bennett S J. – Inherited Disorders of Platelet Function - ASH/2005.")
85
hematol/Disease-findings.htm disorders_feature. faculty.washington.edu/ kepeter/118/photos/blo... pages/newpage20.htm

86
Obrigada pela Atenção ! Nydia Strachman Bacal nsbacal@einstein.br

87
Drogas

88
ISLH Miami - 2007 – Aula Dra K.Kottke-Marchant

94
Outros agonistas Epinefrina: trifásica (plaqueta em repouso, agregação primária, agregação secundária)
")
97
Síndrome de Bernard Soulier - Fisiopatologia
Defeito na adesão das plaquetas ao FvW Sítio de Ligação com o FvW e a Trombina na GpIba Formação de macroplaquetas e trombocitopenia Adesão com citoesqueleto e proteínas sinalizadoras

98
Como a plaqueta promove a hemostasia

99
Diagnóstico Tempo de Sangramento prolongado (> 20 min), trombocitopenia (plaqueta <20.000/mm3), esfregaço periférico apresenta plaquetas gigantes (diâmetro médio >3.5 microns)
, trombocitopenia (plaqueta <20.000/mm3), esfregaço periférico apresenta plaquetas gigantes (diâmetro médio >3.5 microns)")
100
Diagnóstico Agregação plaquetária normal com todos os agonistas, exceção com a Ristocetina (oposto da Trombastenia) Citometria de Fluxo: diminuição da expressão antigênica para a GPIb – CD42b GPIX – CD42a GPV – CD42d

101
O anticorpo CD42b é diretamente ligado a glicoproteina plaquetária GPIb
Essa glicoproteina plaquetária atua como receptor para o Fator Von Willebrand e apresenta uma alta afinidade ao receptor de trombina. Raramente utilizamos o CD42a(GPIX) e o CD42d(GPV) . O antígeno está ausente ou presente em baixa expressão em pacientes com Síndrome de Bernard-Soulier . CD41 (GPIIb) and CD61 (GPIIIa) – Trombastenia de Glanzmann
 e o CD42d(GPV) . O antígeno está ausente ou presente em baixa expressão em pacientes com. Síndrome de Bernard-Soulier . CD41 (GPIIb) and CD61 (GPIIIa) – Trombastenia de Glanzmann.")
102
PFA-100® Platelet Function Analyzer
Bleeding Time - Ivy

103
TS x PFA-100 Sensibilidade:
Baixa sensibilidade para triagem de pacientes com sangrameno mucocutâneo Sensibilidade: TS 36% (D. do Pool de Armazenamento) PFA % (D. Von Willebrand) Combinação 48% Boa especificidade Especificidade: TS 91% PFA-100 (Epinefrina) 84% PFA-100 (ADP) 87% Thromb Haemost 2004:04:892 Thromb Haemost 2003:90:483
 PFA % (D. Von Willebrand) Combinação 48% Boa especificidade. Especificidade: TS 91% PFA-100 (Epinefrina) 84% PFA-100 (ADP) 87% Thromb Haemost 2004:04:892. Thromb Haemost 2003:90:483.")
Apresentações semelhantes