Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Dinâmica Molecular Semi- Quântica Com Aplicações Em Moléculas Diatômicas Rodrigo Alves Dias Setembro de 2003

2
UFMG – ICEx – Pós-graduação (Mestrado) Prof. Orientador: Bismarck Vaz da Costa. Prof. Coorientador: Edimar Pesquisador e apresentador: Rodrigo Alves Dias. (Calouro). Órgão financiador: CNPQ.
. Órgão financiador: CNPQ..")
3
Objetivos: Desenvolver programa capaz de simular Dinâmica Molecular Semi- Quântica (DMSQ). Aplicar a DMSQ em Moléculas Simples: H 2, N 2, O 2, CO e H 2 O. Obtendo os modos normais de oscilação.

4
Introdução: Descrição do problema molecular (Elétrons, Núcleos). Aproximação Adiabática (Born-Oppenheimer). Movimento Eletrônico. (Tratado Quântico) Movimento Nuclear. (Tratado Clássico) Forças sobre os núcleos. Evolução Temporal Nuclear.
. Movimento Eletrônico. (Tratado Quântico) Movimento Nuclear. (Tratado Clássico) Forças sobre os núcleos. Evolução Temporal Nuclear..")
5
Problema Molecular:

6
Principio Variacional: Dada a Eq. De Schrödinger dependente do tempo: Supondo, Eq. De Schrödinger independente do tempo. Descrição dos estados estacionários. Definição do funcional energia:

7
Condição de Extremos. Quando, Estados Estacionários do Funcional Energia Supondo, Estados Estacionários: Equação de autovalores Generalizada.

8
Aproximação de Born-Oppenheimer: Sabemos que, M A > m e (M p,n =1836,15*m e ), logo podemos supor que os núcleos são muito mais lentos que os elétrons. Portanto, uma boa aproximação é considerar os núcleos fixos.

9
Definindo, Fornecendo um potencial do tipo molecular!

10
O Movimento Eletrônico Hartree-Fock: Uma solução aproximada é necessária devido às muitas interações! Assim um elétron será descrito por uma função de onda que se estende por toda a molécula, chamada Orbital Molecular(O.M.) denotado por: designa um certo elétron. i designa os vários orbitais moleculares. Spin Orbital Molecular (S.O.M.). Princípio da Exclusão de Pauli. Anti-Simetria
 denotado por: designa um certo elétron. i designa os vários orbitais moleculares. Spin Orbital Molecular (S.O.M.). Princípio da Exclusão de Pauli. Anti-Simetria.")
11
Assumindo que: (L.I.) Tomando a condição de extremo ( E elet =0), mantendo a restrição de que os orbitais moleculares sejam (L.I.) e aplicando a técnica dos multiplicadores de Lagrange obtemos: Considerando um sistema de camada fechada onde os S.O.M. são:

12
O Operador Hamiltoniano Hartree-Fock: E dado que, Matriz dos multiplicadores de Lagrange é hermitiana. Se existe U tal que, As melhores O.M. satisfazem.

13
Seus autovalores são reais; Auto funções pertencentes a diferentes autovalores são ortogonais; Os n autovalores mais baixos de F, os E i, e as correspondentes autofunções i forneceram uma descrição do estado fundamental do sistema. As demais são apropriadas para uma descrição de estados excitados; Procedimento Geral de Solução de : Dado { i ’ } geral, pela minimização da energia, um novo conjunto de { i ’’ } é encontrado ate que { i ’’ }- { i ’ }= . critério de convergência.

14
O Procedimento LCAO: Dado que, Matriz de Superposição (overlap)., onde os ´ s são orbitais atômicos. Em geral, Utilizando a condição de extremo ( E=0) e aplicando a técnica dos multiplicadores de Lagrange obtemos que:
 e aplicando a técnica dos multiplicadores de Lagrange obtemos que:.")
15
O conjunto de equações satisfeitas pelos melhores C i ’ s são as Equações de Hartree-Fock-Hoothann: Existem n equações deste tipo, uma para cada O.M., a solução de cada uma delas fornecerá o conjunto de coeficientes (C i ’ s) de cada orbital molecular na base dos orbitais atômicos( ) juntamente com a energia( i ) associada a cada um dos orbitais moleculares. Pode-se mostrar, que os autovalores destas equações são as raízes da equação secular: Raízes reais Autovetores Ortogonais Estado Fundamental n soluções i e C i com os autovalores mais baixos ocupados dois a dois.

16
Processo interativo na solução para determinar os melhores coeficientes. Calculo das integrais,, e Calculo dos F Resolução de (F- S)C=0 Calculo dos P Calculo de P = P i - P i+1 se | P | > Cálculo dos novos F se | P | Convergência i N o de interações. critério de convergência.
C=0 Calculo dos P Calculo de P = P i - P i+1 se | P | > Cálculo dos novos F se | P | Convergência i N o de interações. critério de convergência..")
17
Solução Clássica do Problema Nuclear-Dinâmica: Dado que conhecemos, elet definiremos o hamiltoniano clássico por: Usando as equações canônicas de Hamilton obtemos:

18
Utilizamos o método Leap-Frog definido por: Integração Numérica: Com erro, =O(h 2 ). Forças de Helmann-Feyman Correções de Pulay

19
Resultados da DMSQ em algumas Moléculas Simples: 1 o Sistema de Interesse: H 2 Estado Fundamental NHNH 1s NHNH NHNH NHNH Calculamos, Integramos, as equações de movimento e obtemos: Usando o programa GAMESS.

20
H 2 Estado Fundamental

21
H 2....

22
OrbitalOrbital: Animações H 2 :

23
CO Estado Fundamental

24
Animação COAnimação CO: CO....

25
O 2 Estado Fundamental

26
Animação O2Animação O2: O 2....

27
N 2 Estado Fundamental

28
N 2.... Animação N 2 Animação N 2 :

29
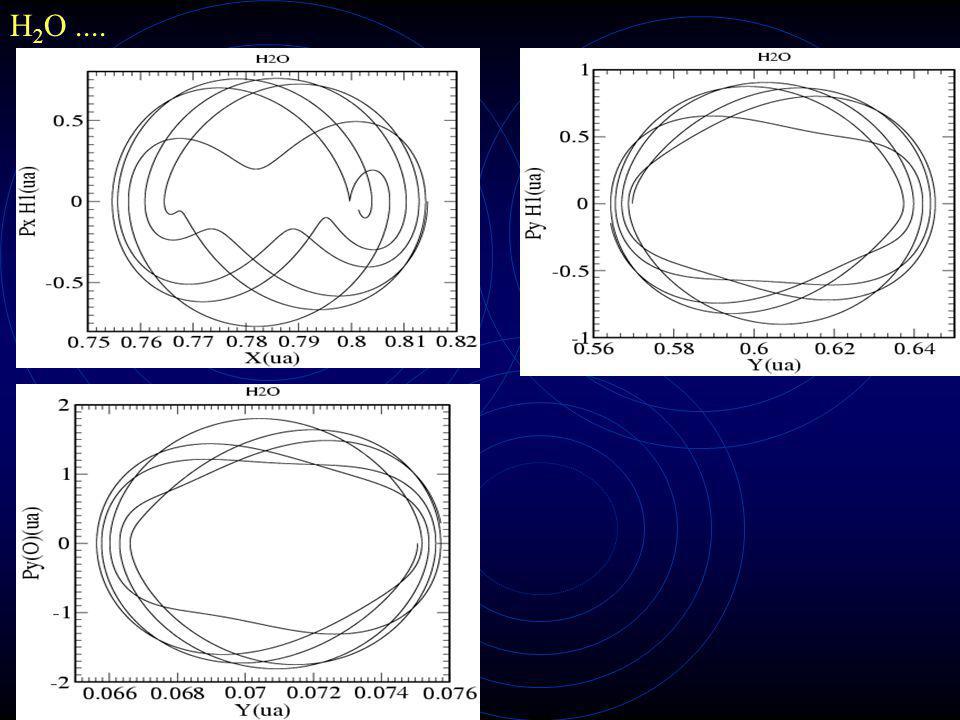
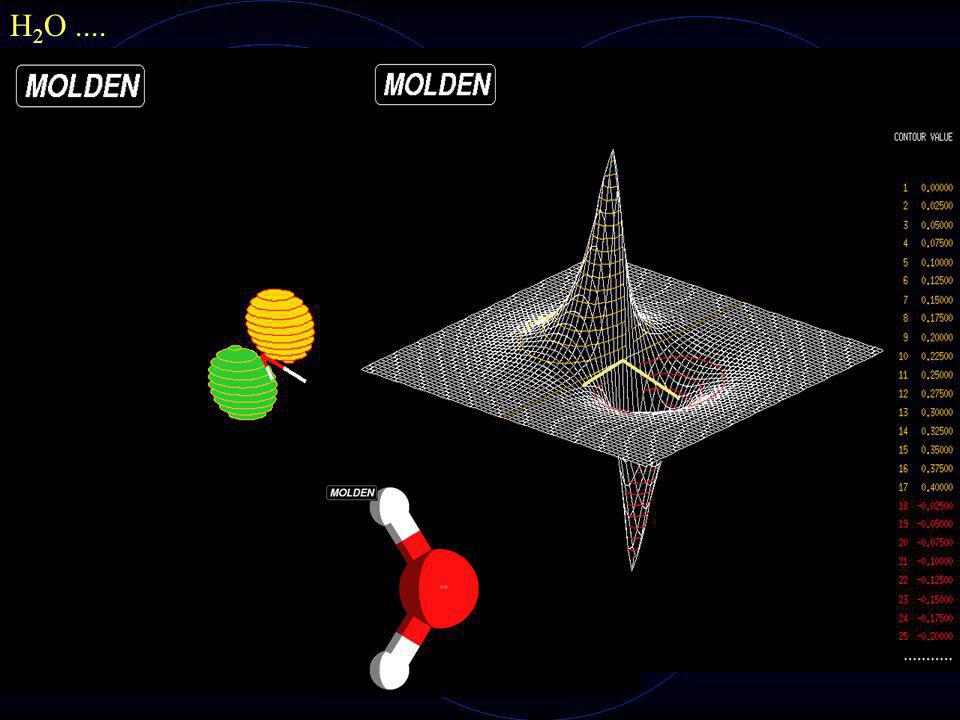
H 2 O Estado Fundamental

30
H 2 O....

33
Conclusões e Perspectivas: Dado o Problema Molecular, utilizando a aproximação adiabática conseguimos separar o movimento eletrônico do movimento nuclear. Movimento eletrônico foi tratado dentro das aproximações de Hartree-Fock-Hoothann (LCAO). Há Interações de Troca. Não há Correlações. (1 det Slater.) Utilizando o GAMESS calculamos a energia eletrônica e as forças que os núcleos sentem. Meu papel foi resolver as equações canônicas clássicas de Hamilton para o movimento dos núcleos e desenvolver o pacote computacional capaz de integrar estas equações numericamente. Obtendo assim a evolução dinâmica do Problema Molecular. Aplicação da DMSQ para o estudo de Superfícies.
. Há Interações de Troca. Não há Correlações. (1 det Slater.) Utilizando o GAMESS calculamos a energia eletrônica e as forças que os núcleos sentem. Meu papel foi resolver as equações canônicas clássicas de Hamilton para o movimento dos núcleos e desenvolver o pacote computacional capaz de integrar estas equações numericamente. Obtendo assim a evolução dinâmica do Problema Molecular. Aplicação da DMSQ para o estudo de Superfícies..")
34
Agradecimentos: Senhor Jesus Cristo! À minha mãe, meu pai, aos meus irmãos Rômulo e Rogério(Em memória). À minha namorada Pricila que sempre esteve comigo, até nos momentos difíceis. À toda a galera do dia a dia, na UFMG. Aos Professores. José Rachid Mohallen(UFMG), Ricardo Vagner(UFMG), Araken Verneck(UCB), Paulo Eduardo de Brito(UCB), Edimar e a todos os outros que porventura eu não tenha citado. Aos irmãos Luiz Gustavo, Cabelo, Philippe, Bráulio, Gabriel. Ao CNPq, que financiou este trabalho. À CBF, pelo Penta Campeonato de 2002. Aos amigo(a)s, Mariana, Mada, Gorginho, Custela, Thiago, Ana Julia, Lets, Para, Schneider, Álvaro(s), Humberto, Caudão, Indhira, Rafael, Clarissa, Gabriela, Mancebo, Alexandre, Miquita, Kajimura, Mauro e outros que porventura eu tenha esquecido.
. À minha namorada Pricila que sempre esteve comigo, até nos momentos difíceis. À toda a galera do dia a dia, na UFMG. Aos Professores. José Rachid Mohallen(UFMG), Ricardo Vagner(UFMG), Araken Verneck(UCB), Paulo Eduardo de Brito(UCB), Edimar e a todos os outros que porventura eu não tenha citado. Aos irmãos Luiz Gustavo, Cabelo, Philippe, Bráulio, Gabriel. Ao CNPq, que financiou este trabalho. À CBF, pelo Penta Campeonato de Aos amigo(a)s, Mariana, Mada, Gorginho, Custela, Thiago, Ana Julia, Lets, Para, Schneider, Álvaro(s), Humberto, Caudão, Indhira, Rafael, Clarissa, Gabriela, Mancebo, Alexandre, Miquita, Kajimura, Mauro e outros que porventura eu tenha esquecido..")
Apresentações semelhantes