Carregar apresentação
1
HEMOFILIA A Giselle Martins Pinto Nelson Gianni de Lima
Rafael Domingos Grando Lisiane Machado Luciana Mendes Johann

2
Introdução Doença de grande interesse médico, científico e público
Freqüente distúrbio hereditário da coagulação Causada por: ausência, deficiência severa ou defeitos na função do fator VIII da coagulação

3
Dados Históricos Relatos antigos presentes no Talmud sugerem seu padrão de herança há aproximadamente 2000 anos “perda de sangue” x “retenção de sangue” Em 1803, Otto descreveu seu padrão de herança numa família de New Hampshire

4
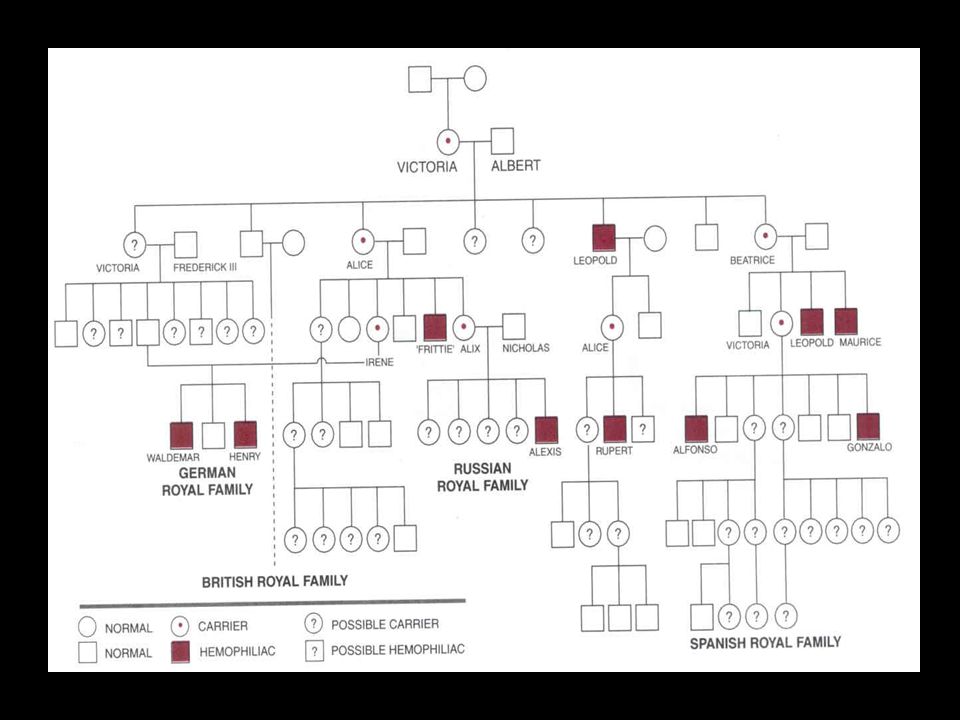
Dados Históricos A Hemofilia ficou conhecida como “doença Real” por seu aparecimento entre os filhos da Rainha Victoria, da Grã-Bretanha Se disseminou às famílias reais européias através dos descendentes da Rainha Victoria Hemofilia A ou Hemofilia B?

6
Definição A hemofilia A é uma doença hereditária recessiva ligada ao cromossomo X que ocorre devido à deficiência do fator VIII da coagulação ou a defeitos estruturais em suas moléculas Distúrbio clinicamente heterogêneo devido à quantidade de defeitos diferentes no gene do fator VIII

7
Epidemiologia É uma doença que não respeita limites étnicos ou geográficos É a principal causa de sangramentos severos e a segunda principal causa de todos os distúrbios congênitos que causam sangramentos, ficando atrás somente da doença de von Willebrand

8
Epidemiologia Incidência aproximada:
- 1 em cada 5000 crianças do sexo masculino - em mulheres, é de 1 para cada

9
Padrão de Herança Doença recessiva ligada ao cromossomo X

10
Padrão de Herança Expressa-se em todos os homens que a recebem, mas apenas nas mulheres homozigóticas para a mutação O homem afetado transmite o gene para todas as suas filhas Mulheres carreadoras têm aproximadamente 50% de chance de transmitir os genes para seus filhos O gene jamais se transmite diretamente do pai para o filho Aproximadamente 30% dos casos decorrem de mutações novas

11
Padrão de Herança Mulheres afetadas: - Homozigóticas recessivas
- Hemizigóticas para a mutação do fator VIII, como resultado de inativação desigual do cromossomo X (defeito na Lyonização) Alguns casos: - Mulheres com síndrome de Turner - Casos de mosaicismo do cromossomo X
 Alguns casos: - Mulheres com síndrome de Turner. - Casos de mosaicismo do cromossomo X.")
13
Apresentação Clínica Ocorre nas seguintes formas
Leve: 31%* Moderada: 26%* Severa: 43%* *Soucie, J. M.; Evatt, B.; Jackson, D.; Hemophilia Surveillance System Project Investigators : Occurrence of hemophilia in the United States. Am. J. Hemat. 59: , 1998.

14
Nível de Fator VIII (ativo)
Apresentação Clínica Classificação Nível de Fator VIII (ativo) Achados Clínicos Severa Até 1% do normal Hemorragias e hemartroses espontâneas, requerindo reposição de fator VIII. Moderada 2-5% do normal Hemorragias secundárias a traumas ou cirurgias. Leve 6-30% do normal Hemorragias secundárias a traumas ou cirurgias, raras hemorragias espontâneas.
 Achados Clínicos. Severa. Até 1% do normal. Hemorragias e hemartroses espontâneas, requerindo reposição de fator VIII. Moderada. 2-5% do normal. Hemorragias secundárias a traumas ou cirurgias. Leve. 6-30% do normal. Hemorragias secundárias a traumas ou cirurgias, raras hemorragias espontâneas.")
15
Hemartrose

16
Hematoma

17
Hematoma do ílio-psoas

18
Outros Achados Pseudotumores Hematúria Complicações neurológicas
Hemorragias em mucosas Sangramentos pós-cirúrgicos

19
Hemorragia Intracraniana

20
Papel do Fator VIII na Coagulação Sangüínea

21
Hemostasia Espasmo Vascular Formação do tampão plaquetário
Formação do coágulo sangüíneo Organização fibrosa Dissolução da crase sangüínea

22
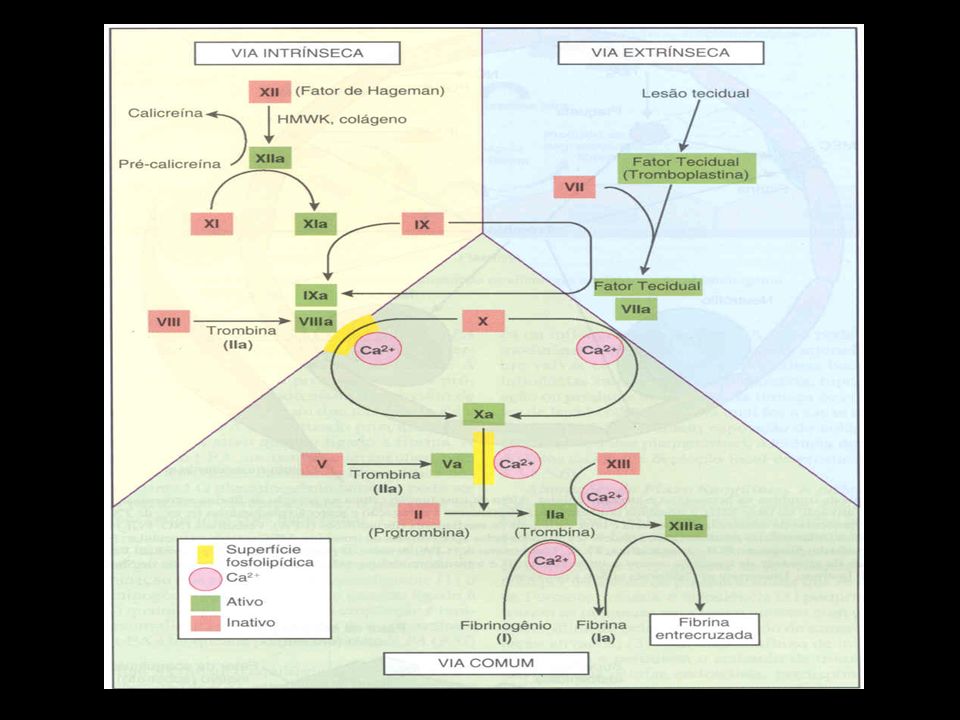
Via Intrínseca Inicia com alteração no sangue ou exposição deste ao colágeno do vaso traumatizado Fator VIII: cofator na via intrínseca da cascata de coagulação

23
Complexos Enzimáticos
Complexo X-ase: fator VIIIa + fator IXa Ativação do fator X Complexo protrombinase ou ativador da protrombina: fator Xa + fator Va

25
Fator VIII Mesmo quando presente em concentrações relativamente baixas no plasma, a função de coagulação é mantida Somente uma redução substancial (maior que 70%) leva aos distúrbios sangüíneos característicos da hemofilia A
 leva aos distúrbios sangüíneos característicos da hemofilia A.")
26
Fator de von Willebrand
Circula no plasma com o fator VIII Funções: acentuar a síntese de fator VIII proteger o fator VIII contra a proteólise aumentar a concentração do fator VIII no sítio da lesão vascular

27
Estrutura do Gene do Fator VIII

28
Estrutura do Gene do Fator VIII
O gene está localizado no topo do braço longo do cromossomo X (Xq28) 186 kb 0,1% da constituição total do X 26 éxons RNAm de 9kb
 186 kb. 0,1% da constituição total do X. 26 éxons. RNAm de 9kb.")
29
Estrutura do Gene do Fator VIII
24 éxons variam em comprimento ( pares de bases) Os maiores éxons são: éxon 14: 3106 pb éxon 26: 1958 pb
 Os maiores éxons são: éxon 14: 3106 pb. éxon 26: 1958 pb.")
30
Estrutura do Gene do Fator VIII
Íntron 22 separa os éxons 22 e 23 Contém a ilha CpG Tem 2 transcrições: F8A e F8B F8B: transcrito na mesma direção que o gene do fator VIII, usando um éxon próprio mais os éxons 23 a 26 do fator VIII F8A: não contém íntrons e é transcrito na direção oposta do gene do fator VIII

31
Estrutura do Gene do Fator VIII
Íntron 22 Há duas cópias de F8A adicionas, não localizadas também no cromossomo X implicadas em quase metade dos casos de hemofilia A severa, via mecanismo de inversão parcial. The Molecular Pathology of Haemophilia A. Review Article.

33
Estrutura do Gene do Fator VIII
RNAm codifica um polipeptídeo de 2351 aa, sendo que 19 aa são removidos durante sua secreção. Hoyer LW. Hemophilia A. N Engl J Med 1994;330:38-47.

34
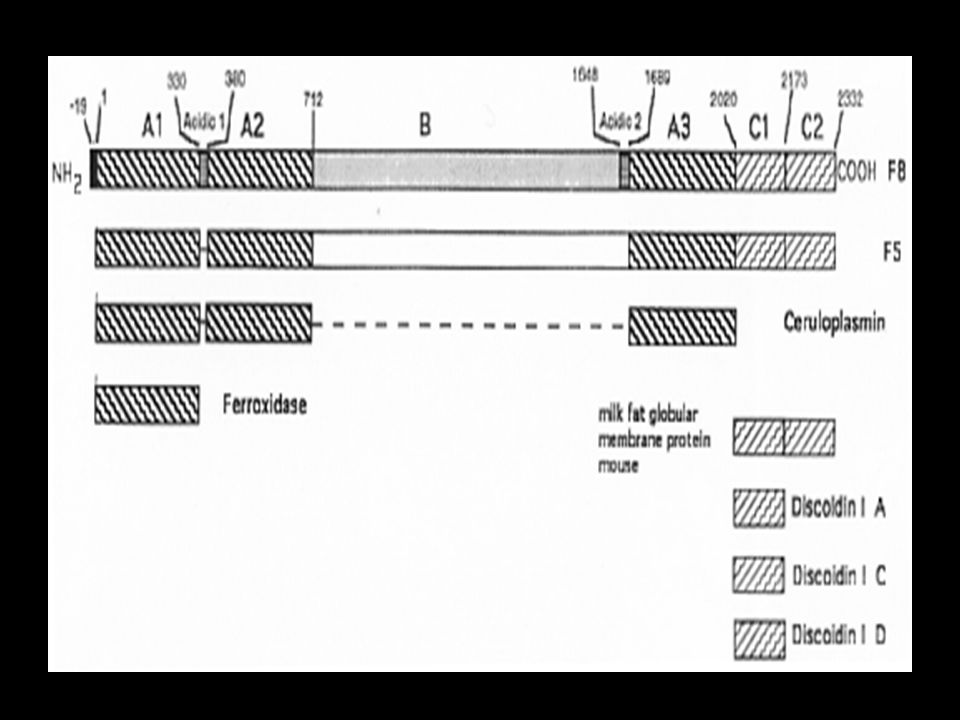
Estrutura do Fator VIII
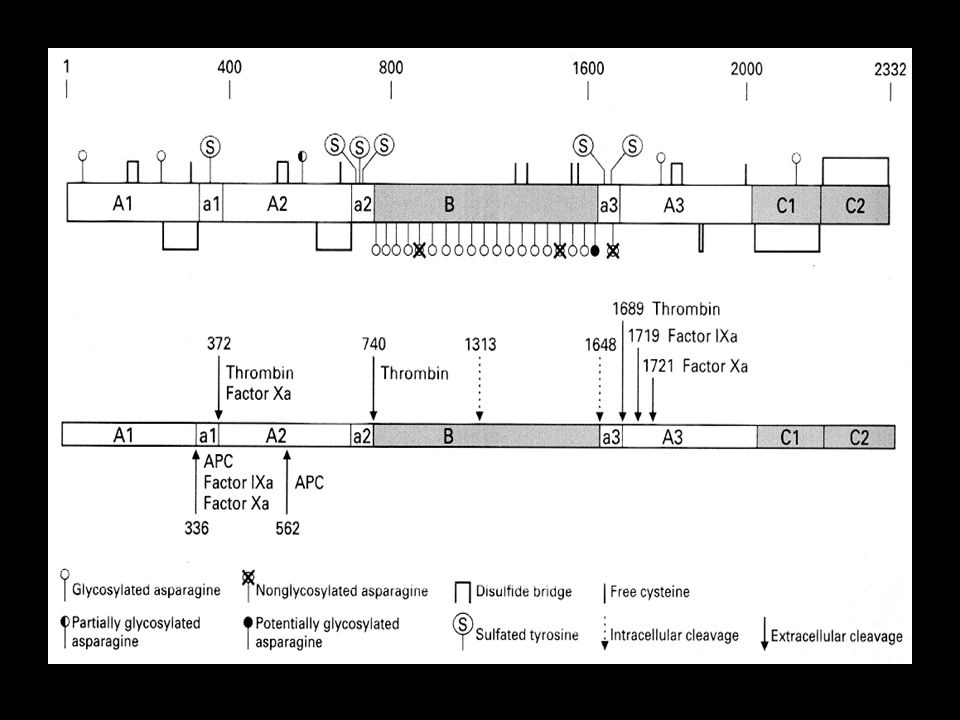
Contém 3 domínios domínio A: A1, A2, A3 domínio B: único e central domínio C: C1, C2

36
Domínio B Codificado pelo éxon 14 Parece exercer a função de conexão
“Fatores VIII recombinantes, cujo domínio B estava ausente, mantinham sua função relativamente normal, mostrando, portanto, que pode ser dispensável.” Hoyer LW. Hemophilia A. N Engl J Med 1994;330:38-47.

37
Domínio B O domínio B é clivado, resultando em :
cadeia leve: A3, C1, C2, de 80 kDa cadeia pesada: A1, A2, de 200 kDa, contém também parte do domínio B

38
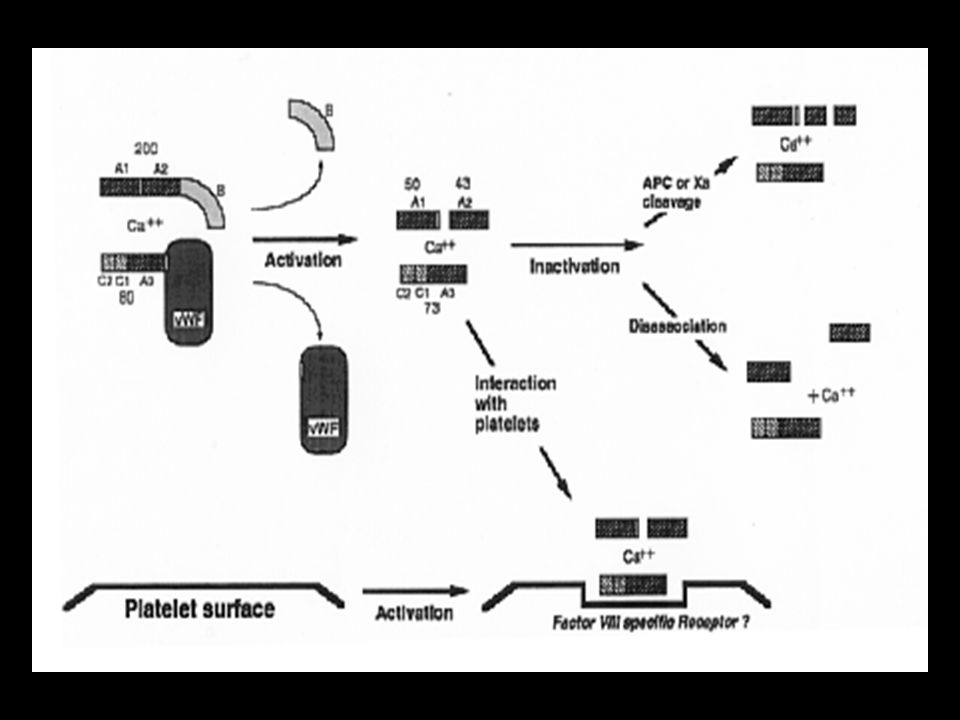
Ativação do Fator VIII Sintetizado como um polipeptídeo de cadeia simples Após a síntese é clivado, por protease ainda indefinida Torna-se um heterodímero

39
Ativação do Fator VIII Pela ação da trombina ou fator Xa, o fator VIII é clivado nos sítios 372 e 1689 da arginina Essa clivagem é essencial para a liberação do fator VIII ligado ao FvW, permitindo que se ligue à superfície fosfolipídica e interaja com o fator IXa

40
Ativação do Fator VIII A clivagem no sítio 372 converte a cadeia pesada em 2 fragmentos: um de 54 kDa e outro de 44 kDa A clivagem no sítio 1689, libera um pequeno fragmento da cadeia leve, separando o fator VIII do FvW. Assim o fator VIII ® fator VIIIa

43
Fator VIIIa É composto pelos seguintes fragmentos:
54 kDa da cadeia pesada 44 kDa da cadeia pesada 72 kDa da cadeia leve A etapa limitante na ativação do fator VIII é a clivagem no sítio 372

44
Fator VIIIa Clivagens não essenciais:
sítio 740 da arginina, pela trombina ou fator Xa a cadeia pesada torna-se um fragmento de 92 kDa retira parte do domínio B da cadeia pesada

45
Desativação do Fator VIII
A função de cofator do fator VIII é rapidamente perdida: Pela proteólise molecular que ocorre por dissociação de suas subunidades. Pela proteína C ativada

47
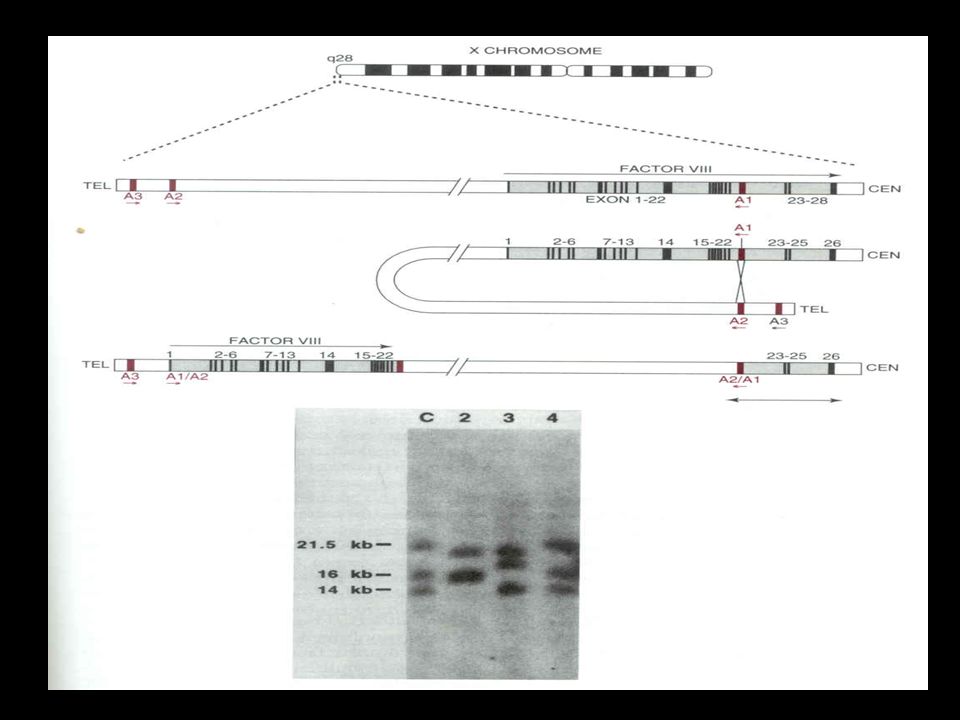
Genética Molecular

48
Genética Molecular Pontos de mutação:
1/3 dos pontos de mutações que ocorre no gene do fator VIII estão localizadas no dinucleotídeo CpG (“ponto quente”) Sítio da endonuclease de restrição Taq1, que reconhece a seqüência TCGA. Essas mutações são detectadas pela perda deste sítio de clivagem. The Molecular Pathology of Haemophilia A. Review Article.
 Sítio da endonuclease de restrição Taq1, que reconhece a seqüência TCGA. Essas mutações são detectadas pela perda deste sítio de clivagem. The Molecular Pathology of Haemophilia A. Review Article.")
49
Defeitos Genéticos Rearranjos gênicos Substituição de Base Única
missense: substituição de um único aminoácido nonsense ou mutação em parada: término prematuro na cadeia peptídica Deleções Grandes deleções Pequenas deleções Inserção

50
Rearranjos Gênicos Consistem basicamente em uma única inversão responsável por mais de 40% de todos os casos de hemofilia A severa. Grande inversão e translocação dos éxons 1 a 22, juntamente com os íntrons, com os éxons 23 a 26 Mecanismo: recombinação homóloga entre F8A e uma cópia extragênica de F8A

51
Rearranjos Gênicos Conseqüência: remoção da terminação C da proteína codificada pelos éxons 23-26, ocasionando uma perda completa na função deste fator Detecção: Southern blotting (enzima de restrição BclI e gene A como probe)
")
53
Rearranjos Gênicos 35% dos casos de hemofilia A severa é pela inversão com a cópia distal de F8A 7% dos casos de hemofilia A severa é pela inversão com a cópia proximal de F8A The Molecular Pathology of Haemophilia A. Review Article.

54
Rearranjos Gênicos Origem: quase exclusivamente é proveniente de um único erro no crossing-over durante a meiose espermatogênica de um homem normal Conclusão: Gera mulheres carreadoras desta mutação, mas sem história familiar de hemofilia A mais de 40% dos casos de hemofilia A severa não têm história familiar

55
Substituição de Base Única
Há aproximadamente 309 tipos 85% são mutações missense 14% são mutações nonsense 1% o splicing do RNAm está alterado ou ausente Nos “pontos quentes” o códon do aa arginina (CGA, CGC, CGG) é freqüentemente afetado por mutações
 é freqüentemente afetado por mutações.")
56
Substituição de única base
Missense: fenótipos variados Nonsense: levam a hemofilia A severa, por formar uma molécula de fator VIII truncado

58
Deleções 5% dos casos de hemofilia
Frequëntemente associado à hemofilia A severa Ocorre tanto nas células germinativas femininas quanto nas masculinas

59
Grandes Deleções Deleções desde 1kb até mais de 210 kb
Mecanismo mais provável: recombinação não-homóloga dos cromossomos na meiose I 5% dos casos de hemofilia A severa Doença moderada: resultado do splicing do RNAm que deleta o éxon 22 e os éxons 23-24 Alto risco para desenvolvimento de inibidores.

60
Pequenas Deleções Variam desde 1 a 86 pares de bases distribuídos pelos éxons. Associadas a doença severa e moderada A maioria são mutações do tipo frameshift

61
Inserções Variam em tamanho de 1 par de bases a 2,1 kb
Todas associadas a doença severa Inserção L1 (LINE 1) similaridade ao DNA da transcriptase reversa retroviral
 similaridade ao DNA da transcriptase reversa retroviral.")
62
Diagnóstico Clínico Sangramentos Hemorragias espontâneas Articulações
Músculos TGI Hemorragias espontâneas

63
Diagnóstico Laboratorial
TTPA Plaquetas, TP, tempo de sangramento normais FvW normal

64
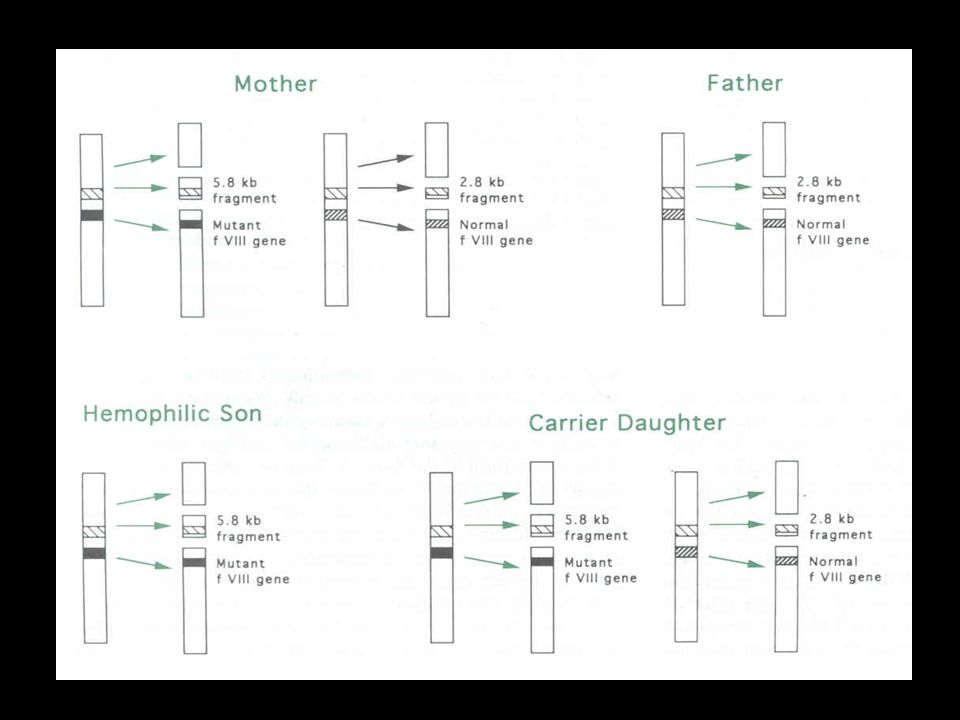
Diagnóstico Genético Mutações de base única PCR + seqüenciamento
Inversões Southern blotting (probe F8A) Deleções Grandes deleções Southern blotting Pequenas deleções PCR + seqüenciamento Inserções Southern blotting
 Deleções. Grandes deleções Southern blotting. Pequenas deleções PCR + seqüenciamento. Inserções Southern blotting.")
65
Diagnóstico Diferencial
Hemofilia B Deficiência de fator IX Doença de von Willebrand FvW deficiente ou anormal Tempo de sangramento prolongado Padrão autossômico dominante

66
Aconselhamento Genético
História familiar Exames laboratoriais: PCR Southern blotting Predisposição à formação de inibidores

67
Aconselhamento Genético
PCR + Seqüenciamento Mutações de base única Dificuldades Grande tamanho da região codificadora Não detecta rearranjos onde a seqüência de éxons esteja inalterada Pequenas deleções Southern blotting Grandes deleções Inserções Inversões

68
Diagnóstico Pré-Natal
Por quê? Assistência psicológica Assistência hospitalar Complicações Determinação do sexo análise citogenética 1979: fetoscopia assistida por ECO = sangue fetal

69
Diagnóstico Pré-Natal
Amniocentese 12-14ª semana de gestação Vilosidades Coriônicas 9-11ª semana de gestação transcervical x transabdominal

70
Tratamento Restituição da atividade deficiente Manejo clínico
centros especializados equipes multidisciplinares avaliações periódicas programas de exercícios boa higiene oral

71
Derivados plasmáticos X Fator VIII recombinante
Tratamento Derivados plasmáticos X Fator VIII recombinante

72
Tratamento Derivados Plasmáticos Anos 70
Crioprecipitado / Plasma fresco Transmissão de agentes infecciosos Processos de inativação viral Formação de inibidores Profilaxia: 3 vezes/semana

73
Tratamento Fator VIII Recombinante Início da década de 90
Excelente eficácia Alta correlação entre dose X níveis fator VII Meia-vida: 8-12 horas Profilaxia: 3 vezes/semana

74
Derivados Plasmáticos Fator VIII Recombinante
Tratamento Derivados Plasmáticos Fator VIII Recombinante segurança? (inativação viral) + seguros + baratos + caros (2-3 vezes) disponibilidade maior baixa disponibilidade inibidores: 10-15% + utilizados
 + seguros. + baratos. + caros (2-3 vezes) disponibilidade maior. baixa disponibilidade. inibidores: 10-15% + utilizados.")
75
Tratamento Anticorpos contra o fator VIII
10-20% dos pacientes hemofílicos Distúrbios severos da crase TTPA prolongado Ac inativam plasma normal

76
Tratamento Schwaab R, Brackmann HH, Meyer C, et al. Haemophilia A: mutation type determines risk of inhibitor formation. Thromb Haemost 1995;74:

77
Tratamento

78
Tratamento Lewis et al. (1985) Tx Hepático
Pcte hemofílico + cirrose por hepatite viral Pós-operatório: níveis quase normais de fator VIII

79
Tratamento Sangramentos Dose
Menores: atingir 25% dos valores normais de fator VIII Moderados (hematomas musculares): atingir 50% Grandes sgtos / cirurgias: atingir 100% Dose 1U fator VIII = 1 ml plasma Volume plasmático = 40 ml/Kg Volume de distribuição do fator VIII = 1,5 vez o volume do plasma 100% 60 unidades/Kg
: atingir 50% Grandes sgtos / cirurgias: atingir 100% Dose. 1U fator VIII = 1 ml plasma. Volume plasmático = 40 ml/Kg. Volume de distribuição do fator VIII = 1,5 vez o volume do plasma. 100% 60 unidades/Kg.")
80
Tratamento Desmopressina Análogo da vasopressina
liberação e níveis de fator VIII e FvW Hemofilia A leve a moderada

81
Doenças Secundárias Fatores plasmáticos (agentes infecciosos)
Cirrose Hepática Vírus da hepatite B, C Ca hepatocelular SIDA HIV Inibidores da protease Trombocitopenia imune Metade da década de 80: 60-70% dos hemofílicos contaminados (EUA e Europa Ocidental)
")
82
Terapia Gênica Candidatas ideais Produção endógena contínua
Vetores virais Introdução do gene em fibroblastos da pele ex vivo

83
Terapia Gênica Roth et al.: Nonviral transfer of the gene encoding coagulation factor VIII in patients with severe hemophilia A. N Engl J Med 2001;344: 4/6 pactes: níveis detectáveis de fator VIII (2 pactes: > 1,0%) Nenhum pcte desenvolveu inibidores ¾ pctes dose de fator VIII recombinante sgtos 10 meses após: indetectáveis
 Nenhum pcte desenvolveu inibidores. ¾ pctes dose de fator VIII recombinante. sgtos. 10 meses após: indetectáveis.")
84
Terapia Gênica Introdução do gene em fibroblastos (eletroforese)
Implantação dos fibroblastos no peritôneo

85
Terapia Gênica

86
Hemofilia A – Realidade
80% dos pacientes não recebem qualquer tipo de tto Produção e purificação do fator VIII extraído do leite de animais transgênicos