Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Renovação de Registro LEI N° 6360/76 decreto 8077 de 2013 Art. 12 ...
§ 1º O registro a que se refere este artigo terá validade por 5 (cinco) anos e poderá ser revalidado por períodos iguais e sucessivos, mantido o número do registro inicial. Revalidação automática ; Renovação data a data 12 a 6 meses antes de vencer o registro.
 anos e poderá ser revalidado por períodos iguais e sucessivos, mantido o número do registro inicial. Revalidação automática ; Renovação data a data 12 a 6 meses antes de vencer o registro.")
2
Renovação de Registro Comprovação de comercialização (por FF e apresentação) PSUR – Periodic Safety Update Report Laudos CQ Importador Listagem de Alterações Pós-Registro Estudos de Estabilidade 4. Todas as empresas, no primeiro semestre do ultimo ano do qüinqüênio de validade do registro já concedido, deverão apresentar os seguintes documentos para efeito de renovação à ANVISA: a) Formulário de petição devidamente preenchido; b) Comprovante de pagamento da taxa de fiscalização de vigilância sanitária ou comprovante de isenção, quando for o caso; c) Certificado de Responsabilidade Técnica, atualizado, emitido pelo Conselho Regional de Farmácia. d) Apresentar documento comprobatório de venda no período de vigência do registro, e os números das notas fiscais e a relação de estabelecimentos compradores em um máximo de 3 (três) notas por forma farmacêutica. Poderá ser apresentada uma declaração referente às apresentações comerciais não comercializadas para as quais a empresa tenha interesse em manter o registro, desde que pelo menos uma apresentação daquela forma farmacêutica tenha sido comercializada. Os Laboratórios Oficiais, quando não houver a produção do medicamento no referido período, apresentar uma justificativa da não comercialização. e) A última versão de bula impressa que acompanha o produto em suas embalagens comerciais. f) Listagem que contemple todas as alterações e/ou inclusões pós-registro ocorridas durante o último período de validade do registro do produto, acompanhados de cópia do D.O.U., ou na ausência cópia do protocolo da(s) petição(ões) correspondente(s); g) Para produtos importados apresentar os respectivos laudos de três lotes importados nos últimos três anos do controle de qualidade físico-químico, químico, microbiológico e biológico, de acordo com a forma farmacêutica, realizado pelo importador no Brasil. h) Dados relativos aos estudos de fase IV se houver. i) Dados de farmacovigilância de acordo com o modelo PSUR/ICH. Estes dados poderão ser requisitados pela ANVISA antes dos prazos de renovação. 2
 Formulário de petição devidamente preenchido; b) Comprovante de pagamento da taxa de fiscalização de vigilância sanitária ou comprovante de isenção, quando for o caso; c) Certificado de Responsabilidade Técnica, atualizado, emitido pelo Conselho Regional de Farmácia. d) Apresentar documento comprobatório de venda no período de vigência do registro, e os números das notas fiscais e a relação de estabelecimentos compradores em um máximo de 3 (três) notas por forma farmacêutica. Poderá ser apresentada uma declaração referente às apresentações comerciais não comercializadas para as quais a empresa tenha interesse em manter o registro, desde que pelo menos uma apresentação daquela forma farmacêutica tenha sido comercializada. Os Laboratórios Oficiais, quando não houver a produção do medicamento no referido período, apresentar uma justificativa da não comercialização. e) A última versão de bula impressa que acompanha o produto em suas embalagens comerciais. f) Listagem que contemple todas as alterações e/ou inclusões pós-registro ocorridas durante o último período de validade do registro do produto, acompanhados de cópia do D.O.U., ou na ausência cópia do protocolo da(s) petição(ões) correspondente(s); g) Para produtos importados apresentar os respectivos laudos de três lotes importados nos últimos três anos do controle de qualidade físico-químico, químico, microbiológico e biológico, de acordo com a forma farmacêutica, realizado pelo importador no Brasil. h) Dados relativos aos estudos de fase IV se houver. i) Dados de farmacovigilância de acordo com o modelo PSUR/ICH. Estes dados poderão ser requisitados pela ANVISA antes dos prazos de renovação. 2.")
3
Pós-Registro de Medicamentos
Lei N° 9782/99 Art. 41. O registro dos produtos de que trata a Lei n° 6.360/76, poderá ser objeto de regulamentação pelo MS e pela Agência visando a desburocratização e a agilidade nos procedimentos, desde que isto não implique riscos à saúde da população ou à condição de fiscalização das atividades de produção e circulação.

4
Pós-Registro de Medicamentos
Resolução RDC n° 209/05 Art. 1º O resultado das análises ... que não implique em modificação no número de registro, será averbado no respectivo ato de registro e divulgado no site da ANVISA.

5
Pós-Registro de Medicamentos
RESOLUÇÃO RDC N° 48, DE 6/10/2009 Art. 3º Este Regulamento aplica-se a medicamentos específicos, genéricos, novos e similares já registrados.

6
RDC 48/09 - Pós-Registro Art. 5º ... implementação imediata, mediante protocolo de petição ou anotação no Histórico de Mudanças do Produto... : I. Alteração ou inclusão de local de embalagem secundária; II. Alteração ou inclusão de local de embalagem primária; III. Alteração menor do processo de produção;

7
RDC 48/09 - Pós-Registro Art. 5º ...
IV. Alteração ou inclusão de equipamento de embalagem primária; V. Alteração ou inclusão de equipamento com mesmo desenho e princípio de funcionamento; VI. Inclusão de tamanho de lote em até 10 vezes; VII. Alteração menor de excipiente;

8
RDC 48/09 - Pós-Registro Art. 5º ...
VIII. Adequação de especificações e métodos analíticos a compêndio oficial ou estreitamento de faixa de especificação; IX. Exclusão de local de fabricação do fármaco ou local de embalagem primária ou local de embalagem secundária ou local de fabricação do produto; X. Redução do prazo de validade com manutenção dos cuidados de conservação.

9
RDC 48/09 - Pós-Registro Art. 16. Medicamentos específicos, não será necessário o envio dos documentos a seguir: I. Relatórios técnicos de estudo de BDR/BE; II. Relatório de perfil de dissolução comparativo.

10
RDC 48/09 - Pós-Registro Art. 18 Nos casos em que a solicitação pós-registro se referir a mais de uma concentração de uma mesma forma farmacêutica, a mesma deverá ser protocolizada com relatório de estabilidade, relatório de produção e laudo de controle de qualidade referente à maior e menor concentração.

11
RDC 48/09 - Pós-Registro Art. 8º O Histórico de Mudanças do Produto - Anexo III deverá ser protocolizado na Anvisa, sendo dispensada a apresentação de Formulários de Petição - FP1 e FP2 para o mesmo e poderá ser objeto de auditoria. IN Nº 11, de 06/10/09

12
Histórico de Mudanças do Produto
Cabeçalho Número do processo Nome Comercial Princípio Ativo Formas Farmacêuticas Concentrações Apresentações1 Período 2 Houve alteração? ( ) Sim ( ) Não Histórico de Mudanças do Produto RDC 48/09 - Anexo III Pós-Registro2 Número do expediente e data de protocolo3 Apresentações envolvidas na mudança Justificativa / descrição / razão da mudança4 Data da aprovação e efetivação da mudança5 Anexo referente a mudança6 1. Informar todas as apresentações registradas; 2. Período a que se refere o Histórico de Mudanças do Produto no formato: "mm/aaaa a mm/aaaa"; 3. Nome do assunto, segundo a norma vigente, preenchido de acordo com a ordem cronológica da efetivação da mudança; 4. Nos casos em que houve protocolização da mudança informar, neste campo, o respectivo número de expediente e data; 5. A empresa deverá preencher neste campo a justificativa da solicitação contemplando a descrição detalhada e a motivação, incluído o argumento técnico para realização da mudança pós-registro; 6. Informar a data de aprovação e efetivação da mudança proposta. Para solicitações pós-registro de realização imediata informar somente a data da efetivação; 7.Preencher o número do anexo referente aos documentos com os dados gerados em função da mudança de acordo com a norma vigente. O anexo deve conter os seguintes documentos: a. Nos casos em que o pós-registro é reportado apenas no HMP deverá ser anexado todos os documentos exigidos pelo assunto; b. Nos casos em que for solicitado protocolo de estabilidade ou for apresentado na solicitação pós-registro estudo de estabilidade acelerado o estudo de estabilidade de longa duração deverá ser anexado quando concluído.
 Sim ( ) Não. Histórico de Mudanças do Produto. RDC 48/09 - Anexo III. Pós-Registro2. Número do expediente e data de protocolo3. Apresentações envolvidas na mudança. Justificativa / descrição / razão da mudança4. Data da aprovação e efetivação da mudança5. Anexo referente a mudança6. 1. Informar todas as apresentações registradas; 2. Período a que se refere o Histórico de Mudanças do Produto no formato: mm/aaaa a mm/aaaa ; 3. Nome do assunto, segundo a norma vigente, preenchido de acordo com a ordem cronológica da efetivação da mudança; 4. Nos casos em que houve protocolização da mudança informar, neste campo, o respectivo número de expediente e data; 5. A empresa deverá preencher neste campo a justificativa da solicitação contemplando a descrição detalhada e a motivação, incluído o argumento técnico para realização da mudança pós-registro; 6. Informar a data de aprovação e efetivação da mudança proposta. Para solicitações pós-registro de realização imediata informar somente a data da efetivação; 7.Preencher o número do anexo referente aos documentos com os dados gerados em função da mudança de acordo com a norma vigente. O anexo deve conter os seguintes documentos: a. Nos casos em que o pós-registro é reportado apenas no HMP deverá ser anexado todos os documentos exigidos pelo assunto; b. Nos casos em que for solicitado protocolo de estabilidade ou for apresentado na solicitação pós-registro estudo de estabilidade acelerado o estudo de estabilidade de longa duração deverá ser anexado quando concluído.")
13
RDC 48/09 - Pós-Registro - HMP
O HMP deverá abranger as modificações pós- registro que ocorreram no último ano. O HMP também deverá conter os relatórios dos estudos de estabilidade que estavam em andamento durante o período. Não será mais necessário o protocolo de aditamento de estabilidade longa duração concluída para estes casos.

14
RESOLUÇÃO RDC N.º 48, DE 6 DE OUTUBRO DE 2009
Art. 7º Art. 7º Todas as petições de alterações, inclusões, suspensões, reativações, e cancelamentos pós-registro que necessitem de protocolização deverão estar acompanhadas dos seguintes documentos: I. Taxa; II. Formulários de Petição - FP1 e FP2; III. Justificativa da solicitação contemplando a descrição detalhada e o racional da proposta, conforme Anexo I. Descrição da solicitação1 Razão da solicitação2 Declaro que nenhuma mudança, além da acima proposta, será realizada e que as informações constantes no texto de bula e rotulagem serão alteradas de acordo com a solicitação acima descrita e serão realizadas somente após a aprovação por esta ANVISA. Responsável técnico 1. Relato contendo a proposta de alteração solicitada pela empresa 2. Motivação da alteração proposta pela empresa incluindo o argumento técnico para a realização da alteração Quando pertinente a empresa deverá anexar documentação comprobatória da motivação

15
RESOLUÇÃO RDC N.º 48, DE 6 DE OUTUBRO DE 2009
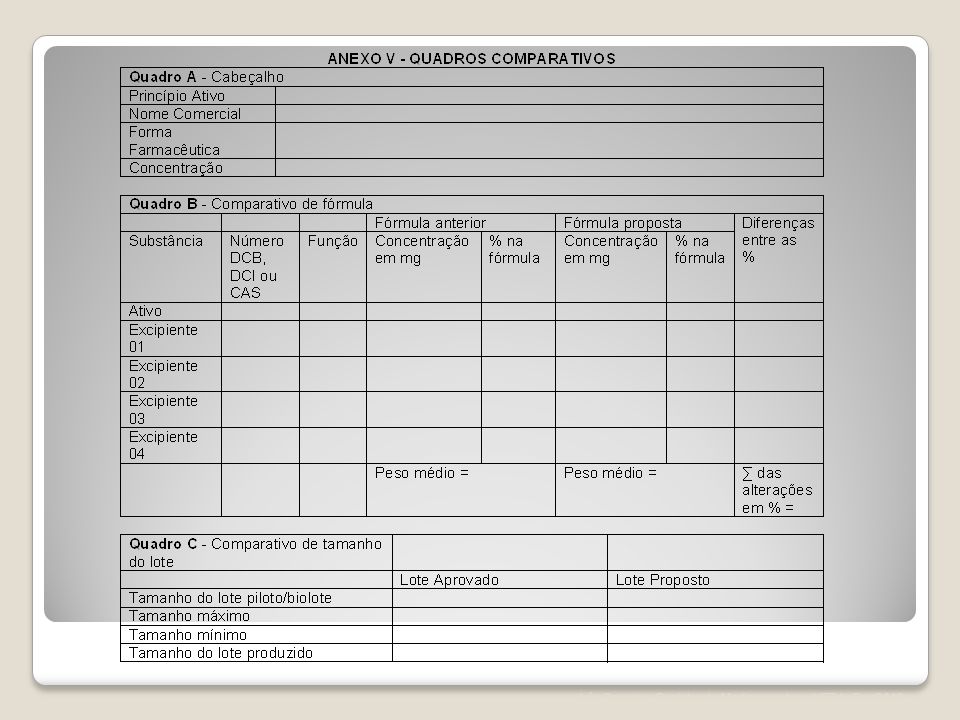
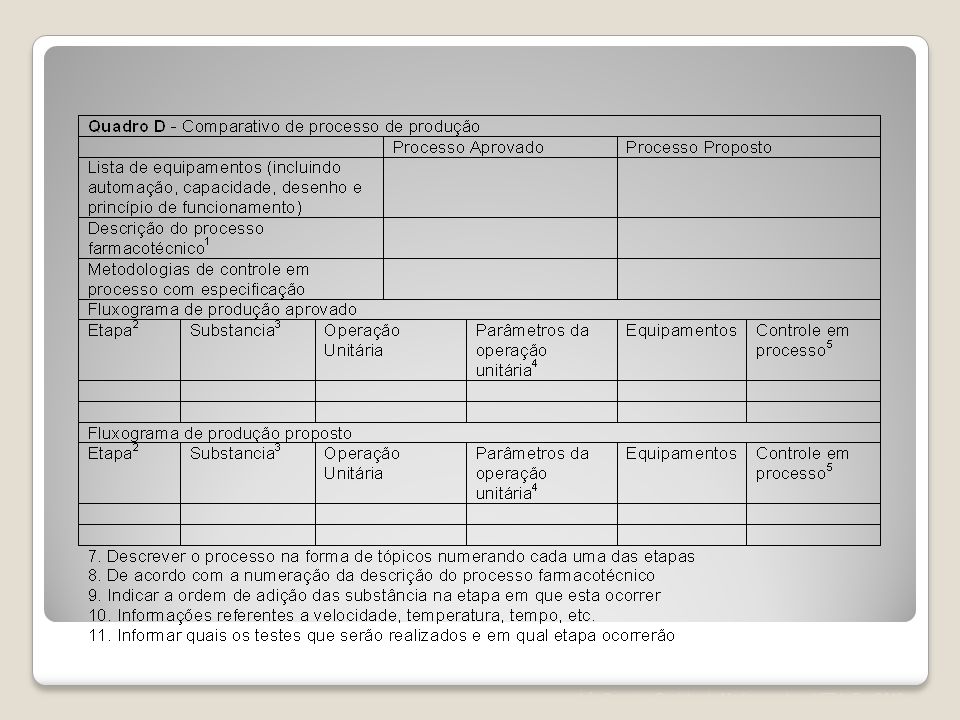
Art. 85. A petição de alteração maior de excipiente deve ser acompanhada dos seguintes documentos: I. Certificado de Boas Práticas de Fabricação válido ou protocolo solicitando a inspeção da Anvisa, desde que a empresa apresente situação satisfatória de acordo com a última inspeção realizada; ou protocolo solicitando a inspeção da Anvisa acompanhado de Certificado de Boas Práticas de Fabricação válido emitido pela autoridade sanitária competente, para os fabricantes internacionais; ou a Anvisa poderá consultar seu banco de dados com o objetivo de comprovar as Condições Técnicas Operacionais da empresa peticionária. II. Relatório de produção, incluindo os quadros comparativos "a" e "b" do Anexo V; III. Especificação e metodologia analítica, com referência bibliográfica utilizada, ou, quando não farmacopeica, descrição da metodologia para os excipientes cujas informações ainda não constem no registro. IV. Informações referentes à encefalopatia espongiforme transmissível, para os excipientes cujas informações ainda não constem no registro; V. Laudo analítico de controle de qualidade do produto acabado referente a 1 (um) lote; VI. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; VII. Relatório de validação do novo método analítico do produto acabado; VIII. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; IX. Relatório de estudo de estabilidade de longa duração referente a 3(três) lotes, a ser incluído no Histórico de Mudanças do Produto; X. Relatório com método e resultados dos testes de eficácia de conservantes, nos casos em que se altera o próprio sistema conservante; XI. Relatório técnico de estudo de biodisponibilidade relativa/ bioequivalência;
 lote; VI. Relatório de perfil de dissolução comparativo entre a condição anteriormente registrada e a nova condição, quando aplicável; VII. Relatório de validação do novo método analítico do produto acabado; VIII. Relatório de estudo de estabilidade referente a 1(um) lote do produto acabado; IX. Relatório de estudo de estabilidade de longa duração referente a 3(três) lotes, a ser incluído no Histórico de Mudanças do Produto; X. Relatório com método e resultados dos testes de eficácia de conservantes, nos casos em que se altera o próprio sistema conservante; XI. Relatório técnico de estudo de biodisponibilidade relativa/ bioequivalência;")
19
RDC 48/09 – Aspectos de Estabilidade
Art. 10. Relatório de estudo de estabilidade = estabilidade acelerada + longa duração em andamento ou estudo de estabilidade de longa duração concluído. Art. 11. Protocolo ou relatório de estudo de estabilidade = dados do estudo de estabilidade a serem incluídos no HMP.

20
RDC 48/09 – Aspectos de Estabilidade
Art. 12. Resultados fora de especificação do estudo de estabilidade em andamento devem ser informados imediatamente à Anvisa e, posteriormente à conclusão da investigação, deverá ser enviada proposta de ação corretiva. Art. 14. Nos casos em que seja solicitado protocolo de estudo de estabilidade, o prazo de validade registrado será mantido.

21
RDC 48/09 – Aspectos de Bula/Rot.
Art. 17 Não será necessário anexar à petição os novos modelos de texto de bula e rotulagem para as alterações pós-registro que necessitem de atualização dos mesmos, exceto quando solicitados.

22
RDC 48/09 – Índice de Assuntos
DAS MUDANÇAS RELACIONADAS AO LOCAL DE FABRICAÇÃO Da Alteração ou Inclusão de local de embalagem secundária Da Alteração ou Inclusão de local de embalagem primária Da Alteração ou Inclusão de local de fabricação do medicamento de liberação convencional Da Alteração ou Inclusão de local de fabricação do medicamento de liberação modificada DAS MUDANÇAS RELACIONADAS AO PROCESSO DE PRODUÇÃO Da alteração ou inclusão menor do processo de produção Da alteração ou inclusão moderada do processo de produção Da alteração ou inclusão maior do processo de produção DAS MUDANÇAS RELACIONADAS AO EQUIPAMENTO Da alteração ou inclusão de equipamento de embalagem primária Da alteração ou inclusão de equipamento com mesmo desenho e princípio de funcionamento Da alteração ou inclusão de equipamento com diferente desenho ou princípio de funcionamento

23
RDC 48/09 – Índice de Assuntos
DAS MUDANÇAS RELACIONADAS AO TAMANHO DO LOTE Da inclusão de tamanho de lote em até 10 vezes Da inclusão de tamanho do lote superior a 10 vezes Art. 66. Refere-se à inclusão de tamanho de lote superior a 10 vezes o tamanho do lote piloto/biolote. DAS MUDANÇAS RELACIONADAS AOS EXCIPIENTES Da inclusão de nova apresentação por alteração de sabor Da alteração menor de excipiente Da alteração moderada de excipiente Da alteração maior de excipiente DAS MUDANÇAS RELACIONADAS À ATUALIZAÇÃO DE ESPECIFICAÇÕES E MÉTODOS ANALÍTICOS DO PRODUTO ACABADO Da adequação de especificações e métodos analíticos a compêndio oficial ou estreitamento de faixa de especificação Da atualização de especificações e métodos analíticos DAS MUDANÇAS RELACIONADAS AO PRAZO DE VALIDADE OU AOS CUIDADOS DE CONSERVAÇÃO Da redução do prazo de validade com manutenção dos cuidados de conservação Da redução do prazo de validade com alteração dos cuidados de conservação Da ampliação do prazo de validade ou alteração dos cuidados de conservação

24
RDC 48/09 – Índice de Assuntos
DA INCLUSÃO DE NOVA APRESENTAÇÃO COMERCIAL Da inclusão de nova apresentação comercial Da inclusão de nova apresentação comercial de produto estéril DA INCLUSÃO DE NOVO ACONDICIONAMENTO DAS MUDANÇAS RELACIONADAS AO FÁRMACO Da alteração ou inclusão da rota de síntese do fármaco Da alteração ou inclusão de local de fabricação do fármaco DAS MUDANÇAS RELACIONADAS À POSOLOGIA Da alteração de posologia Da alteração de posologia para medicamentos específicos DA AMPLIAÇÃO DE USO Da ampliação de uso Da ampliação de uso para medicamentos específicos DA INCLUSÃO DE NOVA VIA DE ADMINISTRAÇÃO Da inclusão de nova via de administração no país Da inclusão de nova via de administração já registrada no país Da inclusão de nova via de administração para medicamentos específicos

25
RDC 48/09 – Índice de Assuntos
DA INCLUSÃO DE INDICAÇÃO TERAPÊUTICA Da inclusão de indicação terapêutica nova no país Da inclusão de indicação terapêutica para medicamentos específicos DA INCLUSÃO DE NOVA CONCENTRAÇÃO Da Inclusão de nova concentração no país Da inclusão de nova concentração já registrada no país Da inclusão de nova concentração para medicamentos específicos DA INCLUSÃO DE NOVA FORMA FARMACÊUTICA Da inclusão de nova forma farmacêutica no país Da inclusão de nova forma farmacêutica já registrada no país Da inclusão de nova forma farmacêutica para medicamentos específicos DAS MUDANÇAS RELACIONADAS À ROTULAGEM DAS MUDANÇAS RELACIONADAS AO NOME COMERCIAL DA SUSPENSÃO TEMPORÁRIA DE FABRICAÇÃO DA REATIVAÇÃO DA FABRICAÇÃO DE MEDICAMENTO

26
RDC 48/09 – Índice de Assuntos
DO CANCELAMENTO DO REGISTRO Do cancelamento de registro da apresentação do medicamento Do cancelamento de registro do medicamento EXCLUSÃO DE LOCAL DE FABRICAÇÃO DO FÁRMACO OU LOCAL DE EMBALAGEM PRIMÁRIA OU LOCAL DE EMBALAGEM SECUNDÁRIA OU LOCAL DE FABRICAÇÃO DO PRODUTO ANEXO I - JUSTIFICATIVA DA SOLICITAÇÃO ANEXO II - ANEXO DE EXCIPIENTES ANEXO III - Histórico de Mudanças do Produto ANEXO IV - RELATÓRIO DE PRODUÇÃO ANEXO V - QUADROS COMPARATIVOS ANEXO VI - RELATÓRIO DE ESTABILIDADE ANEXO VII - MATERIAIS DE ACONDICIONAMENTO

Apresentações semelhantes




















 Liberação paramétrica.>")