Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Validação de Métodos Bioanalíticos Curso de Farmacologia Clínica
Universidade Mogi das Cruzes Março 2004

2
Introdução Nas últimas décadas, houve um aumento de métodos bio-
analíticos aplicados à cromatografia para a determinação qualita- tiva e quantitativa de fármacos, produtos acabados, matérias pri- mas e amostras biológicas, em todas as fases do desenvolvimen- to do fármaco desde a pesquisa até a realização de estudos de bioequivalência. Assim, devido este aumento e a globalização da economia, a adoção de um sistema de qualidade reconhecido universalmente aceito, é requisito fundamental.

3
Portanto, o entendimento do que constituí uma validação
de métodos cromatográficos, possibilita a garantia de um nível satisfatório de qualidade na validação metodológica (Massart et al., 1998; Swartz & Krull, 1998).
.")
4
Desde o final dos anos 80, a importância da validação
de métodos bioanalíticos e suas influências na avaliação e in- terpretação dos resultados, transformou-se em alvo de amplas discussões presentes em diversas Conferências tanto nos EUA quanto na Europa (Shan et al., 1991 e 1992; Dadgar, 1995; Cart- wright et al., 1991 e Arnoux & Morrison, 1992), e consequente- mente através destas, ter melhor direcionamento dos princípios utilizados em bioanálises, tendo com base, principalmente o re- latório da Conferência de Washington de 1992, fornecendo ade- quadas diretrizes em relação aos parâmetros exigidos na valida- ção analítica.
, e consequente- mente através destas, ter melhor direcionamento dos princípios. utilizados em bioanálises, tendo com base, principalmente o re- latório da Conferência de Washington de 1992, fornecendo ade- quadas diretrizes em relação aos parâmetros exigidos na valida- ção analítica.")
5
Para se obter conclusões precisas em Estudos Biofarma-
cêuticos, estas devem ser extraídas a partir de ensaios bioanalíti- cos, onde as concentrações da droga ou seus metabólitos a ser investigados, devem ser exatos, precisos e seletivos (Jackson, J.A 1994), portanto, a correta avaliação dos Ensaios Biofarma- cêuticos só pode ser alcançada se a exatidão dos dados analíti- cos forem obtidos (Pachla et al.,1986). Além disso, essa exatidão depende de critérios fundamentais empregados, não só em rela- ção à adequada interpretação desses resultados, como também na aplicação da confiabilidade e na totalidade do desempenho do método bioanalitico.
, portanto, a correta avaliação dos Ensaios Biofarma- cêuticos só pode ser alcançada se a exatidão dos dados analíti- cos forem obtidos (Pachla et al.,1986). Além disso, essa exatidão. depende de critérios fundamentais empregados, não só em rela- ção à adequada interpretação desses resultados, como também. na aplicação da confiabilidade e na totalidade do desempenho. do método bioanalitico.")
6
Assim, pode-se destacar como principais critérios* :
Avaliação da droga e estabilidade do analito . Seletividade/Especificidade. Linearidade e modelo de calibração. Precisão e Exatidão. Sensibilidade, Limite de Quantificação e Detecção Recuperação * Pachla et al., 1986; Buick et al., 1990; MacDouglas e Crummett, 1980; Taylor, 1983; Brooks e Weinfeld,1985; Dadgar e Smith, 1986; Inman et al., 1987 e Shan et al., 1987.

7
Para uma correta interpretação dos resultados, atualmente
têm-se proposto uma adequação no planejamento e na realiza- ção das fases clínica e analítica compreendidas em Estudos Bio- farmacêuticos. Protocolos clínicos normalmente são desenvolvi- dos utilizando elevado número de dados de informações pré- clínicas e clínicas, a fim de minimizar riscos que os pacientes poderão sofrer. Da mesma maneira, métodos analíticos são utilizados na quantificação de drogas e seus metabólitos em matrizes bioló- gicas, os quais deverão ser cuidadosamente desenvolvidos para otimizar sua precisão e exatidão.

8
Modernos métodos bioanalíticos são fundamentados em
relação a uma variedade de técnicas físico-químicas e biológicas a saber: Métodos Químicos: Cromatografia (CG, HPLC) e uma variedade de processos utilizando métodos de espectrometria de massa (MS) tais como MS-MS e combinações de técnicas tais como CG-MS,LC-MS. Métodos Biológicos: baseados em procedimentos de imunoensaios; em particular o Radioimunoensaio (RIA), imunoensaio de múltiplas enzimas (EMIT) e ensaio de imunoabsorção de enzimas ligadas (ELISA). Métodos microbiológicos
 e uma variedade de processos utilizando métodos de espectrometria de massa (MS) tais como MS-MS e combinações de técnicas tais como CG-MS,LC-MS. Métodos Biológicos: baseados em procedimentos de imunoensaios; em particular o Radioimunoensaio (RIA), imunoensaio de múltiplas enzimas (EMIT) e ensaio de imunoabsorção de enzimas ligadas (ELISA). Métodos microbiológicos.")
9
“São capazes de fornecer precisão e exatidão em relação
aos resultados obtidos (Jackson, J.A 1994)”.
 .")
10
Tais métodos analíticos devem ser cuidadosamente pré-
validados na fase de desenvolvimento e também validados du- rante o ensaio analítico, para garantir que os mesmos sejam sa- tisfatoriamente realizados e que a confirmação das especifica- ções pré-determinadas seja encontrada; além disso, gerar confia- bilidade nos resultados obtidos (Buick et al., 1990; Jackson, J. A 1994; Shan et al., 1992). Para tanto, os mesmos envolvem procedimentos de de- terminação e quantificação de moléculas orgânicas com amplas propriedades físico-químicas, onde cada composto deve ser avaliado de acordo com as suas propriedades individuais e tam-
. Para tanto, os mesmos envolvem procedimentos de de- terminação e quantificação de moléculas orgânicas com amplas. propriedades físico-químicas, onde cada composto deve ser. avaliado de acordo com as suas propriedades individuais e tam-")
11
bém levando em consideração a complexidade analítica e as
concentrações almejadas (Jackson, J. A 1994). A dimensão de como o método é validado, depende da sua aplicação, número de amostras a ser investigadas e como se- rá o seu desempenho em relação aos dados obtidos (Pachla et al., 1986; Buick et al., 1990).
. A dimensão de como o método é validado, depende da. sua aplicação, número de amostras a ser investigadas e como se- rá o seu desempenho em relação aos dados obtidos (Pachla et. al., 1986; Buick et al., 1990).")
12
Diversos métodos analíticos podem ser necessários duran-
te o desenvolvimento de fármacos. Cada método deve ser vali- dado quando é aplicado em diferentes estudos. Neste caso, quando possível, o número de métodos aplica- dos deve ser mínimo para que as informações sejam estatistica- mente avaliadas, quando compiladas em relação às já existentes (Pachla et al., 1986).
.")
13
Parte das técnicas analíticas podem exigir validação
adicional como: Investigação das características do complexo antígeno-anticorpo. Determinação do pico de pureza Avaliação do efeito de matrizes ou conformação estrutural do composto.

14
Normalmente, cada ensaio deve possuir validação cruzada
empregando técnicas com detectores de alta especificidade, por exemplo, o espectrômetro de absorção de massa (Buick et al., 1990). Em uso, o método analítico validado, o seu desempenho deve ser monitorado por padrões de controle de qualidade, para garantir sua aplicabilidade. No caso de um método analítico ser realizado em outro laboratório, o seu desempenho deve ser mo- nitorado através de padrões de controle de qualidade enviados pelo laboratório de origem.
. Em uso, o método analítico validado, o seu desempenho. deve ser monitorado por padrões de controle de qualidade, para. garantir sua aplicabilidade. No caso de um método analítico ser. realizado em outro laboratório, o seu desempenho deve ser mo- nitorado através de padrões de controle de qualidade enviados. pelo laboratório de origem.")
15
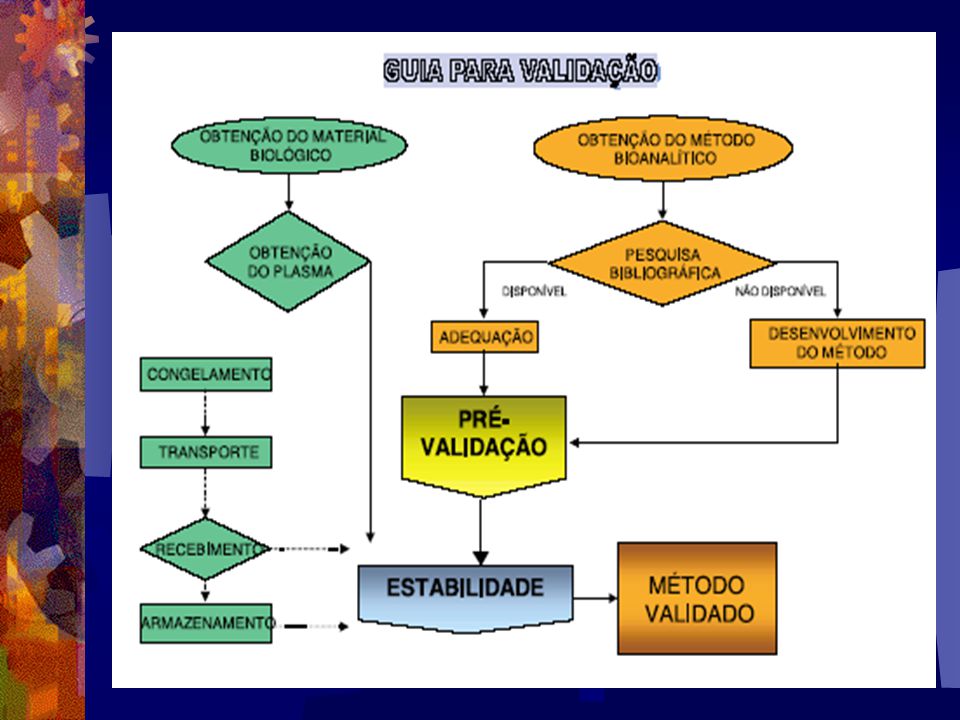
Seleção e Desenvolvimento
Pode-se considerar que a etapa crítica no Estudo Biofarma- cêutico é o desenvolvimento e validação de ensaios bioanalíti- cos, que consistem de experimentos realizados para encontrar condições necessárias para a quantificação do analito em ques- tão. Isto requer conhecimentos das propriedades físico-quími- cas da droga para que o processo de desenvolvimento seja ra- cional e individualizado. Assim, todos os critérios envolvidos no desenvolvimento analítico deverão ser registrados em relató- rios de acompanhamento analítico (Snyder, 1988; Jackson, J.A 1994).
.")
16
A realização de uma pesquisa bibliográfica é a primeira
etapa para a busca do método bioanalítico. Uma vez existindo o método, ele deverá ser testado quanto a sua reprodutibilidade. Na inexistênciade um método bioanalítico para um deter- minado fármaco, o centro analítico deve desenvolver um méto- do que responda satisfatóriamente ao estudo desejado. A realização prévia das etapas necessárias no desenvolvi- mento do método analítico para os estudos de bioequivalência assegura ao centro analítico e ao seu contratante que os serviços contratados serão realizados no tempo previsto e com a confia- bilidade necessária dos resultados, os quais serão avaliados para fins de registro do medicamento em estudo.

17
Neste contexto pode-se afirmar que contrato e contratan-
te não perderão tempo e nem recursos financeiros adicionais se por acaso os estudos realizados forem rejeitados no seu término em função da inadequabilidade do método utilizado e das con- dições de armazenamento não determinadas.

18
Para a aplicação deste fato, consideram-se os seguintes
tipos de validação: Validação Total Validação total é de importância no desenvolvimento e implementação de um método quando aplicado pela primeira vez ou quando for utilizado para determinar um novo analito nesta mesma condição analítica. Validação Parcial Validações parciais são modificações do método já va- lidado. Uma validação parcial pode compreender desde uma pequena determinação de precisão/exatidão a até quase uma validação total.

19
Algumas mudanças típicas no método se enquadram
nesta categoria, mas não são limitadas a: Transferências de método bioanalítico entre laborató - rios ou analistas; Mudança na metodologia analítica (ex. mudança no sistema de detecção); Mudança no anticoagulante na coleta do fluido bioló- gico; Mudança no processamento das amostras;
; Mudança no anticoagulante na coleta do fluido bioló- gico; Mudança no processamento das amostras;")
20
Mudança relevante na faixa de concentração;
Mudanças nos instrumentos e/ou software; Volume de amostra limitado (ex. estudo pediátrico) Demonstração seletiva de um analito na presença de medicações concomitantes.
 Demonstração seletiva de um analito na presença de medicações concomitantes.")
21
No desenvolvimento de um método é necessário verifi-
car toda a metodologia de preparação da amostra, a qual envol- ve os processos de extração, separação, purificação, identifica- ção e quantificação do fármaco na matriz biológica. Para tanto, podemos dizer que nesta fase, temos duas etapas distintas a sa- ber:

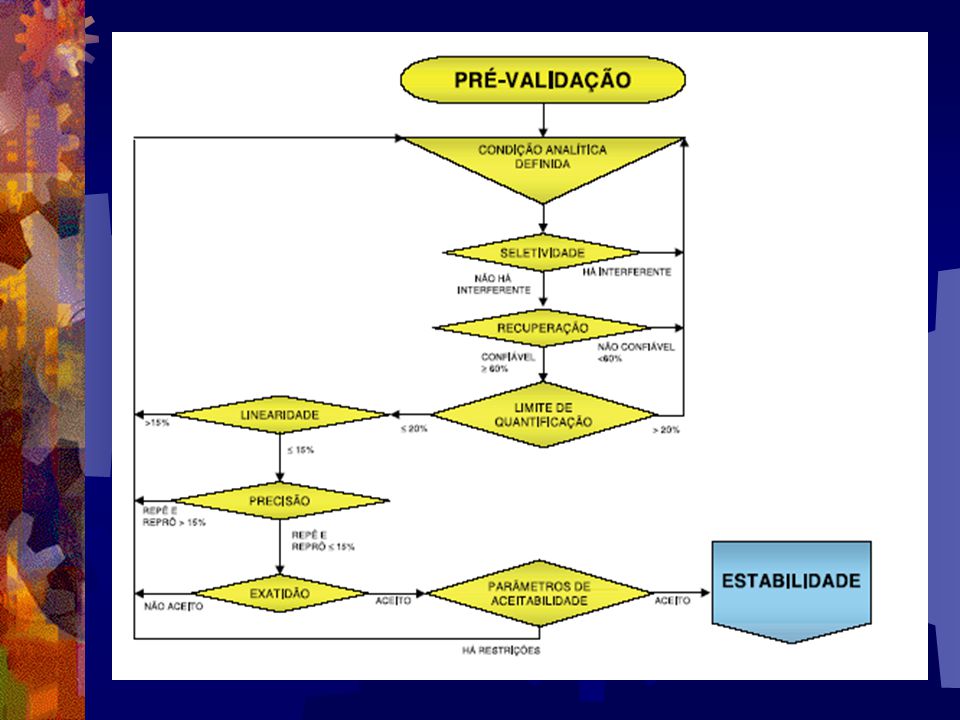
22
Pré-Validação Nesta fase consiste na realização de alguns ensaios preli- minares visando à determinação dos seguintes parâmetros: Exatidão, precisão e recuperação; Linearidade e limites de quantificação; seletividade.

23
Validação do Método É a fase propriamente dita do desenvolvimento e estabe- lecimento da metodologia com o objetivo de definir o ensaio bioanalítico. Neste caso, a padronização de um método bio- analítico típico inclui a determinação dos seguintes parâmetros fundamentais a saber:

24
Especificidade e Seletividade;
Linearidade; Precisão e Exatidão; Sensibilidade, Limite de quantificação e detecção; Recuperação; Estabilidade . Robustez

25
Considerações – Materiais e Reagentes
O uso de substâncias químicas de elevado grau de pureza é fundamental para assegurar a qualidade dos dados analíticos (GARFIELD, 1997; CROSBY et al., 1997). Assim como os rea- gentes químicos utilizados, o uso de SQR é determinante para uma correta quantificação dos fármacos e/ou seus metabólitos. Para uma melhor compreensão deste material, algumas de- finições* são importantes: * Publicações do National Institute of Standards and Technology(NIST) e a Farmacopéia Americana (USP 25) e aceitos pela AOAC (Association of Officials AnalyticalChemists), conforme resumidas por GARFIELD (1997):
. Assim como os rea- gentes químicos utilizados, o uso de SQR é determinante para. uma correta quantificação dos fármacos e/ou seus metabólitos. Para uma melhor compreensão deste material, algumas de- finições* são importantes: * Publicações do National Institute of Standards and Technology(NIST) e a Farmacopéia. Americana (USP 25) e aceitos pela AOAC (Association of Officials AnalyticalChemists), conforme resumidas por GARFIELD (1997):")
26
Padrão primário: de acordo com SKOOG & WEST
(1979), uma substância deve apresentar as seguintes caracterís- cas: deve apresentar elevado grau de pureza, que deve ser determinado; deve ser estável; não deve ser higroscópico ou eflorescente; deve ser de fácil obtenção e preferencialmente de baixo custo; deve apresentar um peso molecular relativamente elevado.
, uma substância deve apresentar as seguintes caracterís- cas: deve apresentar elevado grau de pureza, que deve ser determinado; deve ser estável; não deve ser higroscópico ou eflorescente; deve ser de fácil obtenção e preferencialmente de baixo custo; deve apresentar um peso molecular relativamente elevado.")
27
Material de referência certificado (MRC): material com
uma ou mais propriedades certificadas por procedimentos téc- nicos válidos, acompanhados por ou rastreáveis a um certifica- do ou outro tipo de documentação emitida por um órgão certi- ficador. Material de referência padrão (MRP): material produzi- do pelo NIST. MRPs são certificados em relação a propriedades físico-químicas específicas e acompanhados de certificados que reportam os resultados e indicam o uso do material.
: material produzi- do pelo NIST. MRPs são certificados em relação a propriedades. físico-químicas específicas e acompanhados de certificados que. reportam os resultados e indicam o uso do material.")
28
Padrões de referência USP (USP Reference Standards):
são fármacos purificados ou em elevado grau de pureza, distri- buídos pela USP após recomendação do USP Reference Stan- dards Committee. A seleção dos lotes de matérias-primas utili- zada na preparação destes padrões estão baseadas nas caracterís- ticas críticas de cada fármaco, analisados por três ou mais labo- ratórios, entre os laboratórios da USP, FDA, acadêmicos ou privados (USP 25).
.")
29
Padrão de trabalho ou padrão secundário: preparado a
partir da análise de um lote de material de pureza adequada con- tra um do padrão ou SQR certificado, utilizando metodologia oficial e mantendo o registro das análises. Quando este padrão de trabalho é utilizado na análise de uma amostra, a SQR à par- tir da qual ele foi preparado deve ser mencionada.

30
Além da USP, várias outras farmacopéias, como a Euro-
péia e a Britânica, produzem seus padrões. Estas instituições chamam seus padrões de substâncias químicas de referência (do inglês chemical referece substance, ou simplesmente CRS). Além destas, padrões químicos e biológicos de vários fármacostambém são distribuídos pela Organização Mundial da Saúde (OMS).
. Além destas, padrões químicos e biológicos de vários. fármacostambém são distribuídos pela Organização Mundial da. Saúde (OMS).")
31
Procedimento semelhante foi adotado, recentemente, pela
Farmacopéia Brasileira (F.Bras.) no ano de 2000, o qual foi no- meada a Comissão de Material de Referência (Port. 733, DOU 201-E, de 18/10/00), que iniciou a produção das primeiras substâncias químicas de referência (SQR) da Farmacopéia Brasi- leira. Conforme Resolução RDC nº 56, de 26 de fevereiro de 2002, D.O. de 27/02/2002, após disponí-veis, as SQR da F. Bras. são as SQR oficiais em território nacional e devem ser uti- lizadas obrigatóriamente em relação às demais anteriormente citadas.
 no ano de 2000, o qual foi no- meada a Comissão de Material de Referência (Port. 733, DOU. 201-E, de 18/10/00), que iniciou a produção das primeiras. substâncias químicas de referência (SQR) da Farmacopéia Brasi- leira. Conforme Resolução RDC nº 56, de 26 de fevereiro de. 2002, D.O. de 27/02/2002, após disponí-veis, as SQR da F. Bras. são as SQR oficiais em território nacional e devem ser uti- lizadas obrigatóriamente em relação às demais anteriormente. citadas.")
32
Substâncias químicas de referência (SQR)
O grau de pureza das substâncias químicas utilizadas co- mo referência nos estudos analíticos pode afetar a qualidade dos resultados. O termo SQR refere-se aos padrões do fármaco em estudo e seu(s) metabólito(s), quando for o caso. São substân- cias de elevado grau de pureza, devidamente certificadas. Po- dem ser de dois tipos:
 metabólito(s), quando for o caso. São substân- cias de elevado grau de pureza, devidamente certificadas. Po- dem ser de dois tipos:")
33
SQR disponível comercialmente: sempre que disponíveis,
deverão ser utilizadas SQRs da Farmacopéia Brasileira ou aque- las fornecidas por outras instituições/empresas reconhecidas nacional ou internacionalmente, desde que possibilitem seu rastreamento. SQR não disponível comercialmente: deve ser obtida a partir de substâncias de grau farmacêutico, acompanhado do respectivo certificado de análise do lote e em quantidade sufi- ciente para a produção de um padrão de trabalho que será uti- lizado nos estudos como referência. Este padrão de trabalho somente poderá ser produzido por um Laboratório Analítico Autorizado (LAA), que deverá manter os registros analíticos.
, que deverá manter os registros analíticos.")
34
Existem aqui duas possibilidades para desenvolver o roteiro
de análises para quantificar a matéria-prima como padrão de trabalho: Existência de monografia farmacopéica disponível: nes- te caso o LAA deverá realizar todos os ensaios previstos na mo nografia e emitir um certificado de análise, sendo o teor obtido no ensaio de doseamento o valor de pureza adotado para o padrão de trabalho;

35
Obs.: Caso a monografia não esteja disponível na última edição
da Farmacopéia Brasileira, utilizar preferencialmente as edições mais recentes das farmacopéias Européia, Britânica, Americana ou Portarias do INMETRO. Não existência de monografia farmacopéica: serão admi- tidos estudos com substâncias químicas desde que comprovado sua certificação.

36
Laboratório analítico autorizado (LAA): LAA são os labo-
ratórios REBLAS e Centros Analíticos de Bioequivalência, des- de que apresentem capacidade técnica comprovada para o de- senvolvimento da metodologia analítica indicada.

37
Metabólitos No caso de metabólitos, o centro analítico deverá com- provar, através de certificado de análise do fornecedor ou en- saios realizados no próprio centro, que estes apresentam um grau de pureza definido e adequado para ser utilizado como padrão de trabalho. Padrão Interno Os padrões internos utilizados devem apresentar grau analítico (p.a.) ou superior, de maneira que não interfiram na análise.
 ou superior, de maneira que não interfiram na. análise.")
38
Recomendações: As SQRs, bem como os metabólitos e padrão interno devem ser armazenados conforme instruções do distribuidor. Normalmente, devem ser armazenados em local fresco, ao abrigo da luz e com baixa umidade, sempre em frascos bem vedados. No ato do recebimento deve ser aberta uma cadeia de cus- tódia para cada frasco recebido, na qual se controla o uso da SQR por meio de registro de massa utilizada para cada finali- dade, com visto de quem utilizou. Junto à cadeia de custódia devem ser guardados os certificados de análise das substâncias Deve ser registrado também o fim dado à massa que so- brou da SQR após o vencimento do prazo de validade.

39
Reagentes e solventes Os reagentes e os solventes utilizados nos estudos não devem interferir nos resultados. Isto deve ser verificado através de procedimentos adequados. Devem ser estabelecidos proce- dimentos de controle de fornecedores de maneira a assegurar que solventes e reagentes adquiridos tenham a qualidade dese- jada. Recomenda-se que os fornecedores apresentem certifica- dos analíticos, assim como evidências documentadas para as- segurar a confiabilidade dos mesmos. As áreas de estocagem de substâncias, reagentes, solven- tes e soluções devem ser adequadas.

40
Água grau cromatográfico: deve ter qualidade compatível
com o uso em HPLC. Pode ser: Deionizada Destilada Bi-destilada Ultra-pura A pureza da água deve ser comprovada através de testes adequados.

41
Vidraria A medida precisa de volume é tão importante em muitos métodos analíticos como é a medida de massa. Para tanto, é preciso considerar alguns pontos imprescindíveis para a medi- ção exata de um determinado volume como manutenção dos instrumentos de medição, qualidade dos instrumentos e calibra- ção periódica. As marcas de volume são feitas pelos fabrican- tes com os equipamentos volumétricos bem limpos. Um nível de limpeza análogo deve ser mantido no labo- ratório se estas marcas forem usadas com confiança. Somente superfícies de vidro limpas sustentam um filme uniforme de líquido. Poeira ou óleo rompe este filme. Portanto, a existência de rupturas no filme é uma indicação de uma superfície .suja..

42
O volume ocupado por dada massa de líquido varia com a
temperatura, assim como varia também o recipiente no qual está colocado o líquido, durante a medida. Entretanto, a maioria dos equipamentos de medida de volume é feita de vidro, o qual fe- lizmente tem pequeno coeficiente de expansão. Conseqüentemente, as variações no volume em função da temperatura de um recipiente de vidro não precisam ser consi- deradas em trabalhos em química analítica. As medidas volumé- tricas devem tomar como referência alguma temperatura pa- drão; este ponto de referência é geralmente 20°C. O coeficiente de expansão para líquidos orgânicos pode requerer correções para diferenças de temperatura de 1°C ou até menos, o que tor- na extremamente importante o controle de temperatura am- ambiente dos laboratórios.

43
De uma maneira geral, os procedimentos analíticos são con-
duzidos a uma temperatura que varia entre 15 e 25oC. (British Pharmacopoeia, 2000). A vidraria volumétrica pode ser calibrada individualmente pelo INMETRO, ou laboratório certificado pelo INMETRO. Porém, vidraria .Classe A. satisfaz os padrões internacionais estabelecidos pela International Organisation for Standardiza- tion (British Pharmacopoeia, 2000; GARFIELD,1997;USP 25). O laboratório deve verificar periodicamente os volumes dis- pensados pela vidraria volumétrica, utilizando para isso a massa da água. Para as pipetas, deve também utilizar a massa da água procedendo cinco amostragem para cada volume dispensado (USP2002).
. A vidraria volumétrica pode ser calibrada individualmente. pelo INMETRO, ou laboratório certificado pelo INMETRO. Porém, vidraria .Classe A. satisfaz os padrões internacionais. estabelecidos pela International Organisation for Standardiza- tion (British Pharmacopoeia, 2000; GARFIELD,1997;USP 25). O laboratório deve verificar periodicamente os volumes dis- pensados pela vidraria volumétrica, utilizando para isso a massa. da água. Para as pipetas, deve também utilizar a massa da água. procedendo cinco amostragem para cada volume dispensado. (USP2002).")
44
A exatidão da vidraria .Classe A. é a seguinte:

45
Balanças Balanças (de acordo com Port. 236, de 22 de dez de 1994, do Inmetro) Pesos padrão (de acordo com Port. 233, de 22 de dez de 1994, do Inmetro) As balanças analíticas devem estar instaladas em local adequado, niveladas, livres de correntes de ar, em bancada ex- clusiva para as mesmas e estável. Sempre que possível em sala com temperatura controlada.
 As balanças analíticas devem estar instaladas em local. adequado, niveladas, livres de correntes de ar, em bancada ex- clusiva para as mesmas e estável. Sempre que possível em sala. com temperatura controlada.")
46
A balança deve ser imediatamente limpa após cada uso. Deve
haver um programa de manutenção e conservação da balança, que inclua calibrações periódicas (no mínimo, anualmente), com todas as informações registradas em um livro de registros. Para as balanças analíticas utilizadas em laboratório (Classe I), o valor da divisão real de verificação (d) deve ser de 0,1 mg ou inferior. (Port. 236, de 22 de dez de 1994, do Inmetro). A carga mínima (min) da balança não deve ser inferior a 100 x d. Ex.: Balança analítica com capacidade para 200 g e sensibilidade de 0,1 mg. min = 100 x 0,1 = 10,0 mg
, com. todas as informações registradas em um livro de registros. Para as balanças analíticas utilizadas em laboratório (Classe. I), o valor da divisão real de verificação (d) deve ser de 0,1 mg. ou inferior. (Port. 236, de 22 de dez de 1994, do Inmetro). A carga mínima (min) da balança não deve ser inferior a 100. x d. Ex.: Balança analítica com capacidade para 200 g e sensibilidade. de 0,1 mg. min = 100 x 0,1 = 10,0 mg.")
47
Porém, de acordo com a USP 25 (2002), a incerteza da pesa-
gem (erro sistemático + randômico) não deve ser superior a 0,1% da massa pesada. No caso de balanças eletrônicas que não possuam sistema de auto-calibração, a aferição deve ser feita diariamente, no início do trabalho, e os registros adequadamente armazenados. Os Pesos utilizados devem ser re-certificados anualmente.
 não deve ser superior a. 0,1% da massa pesada. No caso de balanças eletrônicas que não possuam sistema de. auto-calibração, a aferição deve ser feita diariamente, no início. do trabalho, e os registros adequadamente armazenados. Os. Pesos utilizados devem ser re-certificados anualmente.")
48
Equipamentos com temperatura controlada
Refrigerador e freezer deverão ter suas temperaturas veri- ficadas diariamente e registradas no livro de uso. Deve haver um termômetro de máxima e mínima, sendo que a temperatura máxima e mínima do período deverá ser anotada. O local mais adequado para colocar os termômetros é na parte central inter- na do equipamento. Caso a leitura da temperatura seja feita por pares térmicos, estes devem ser calibrados anualmente junto a RBC. Deve haver um POP para refrigerador/freezer descreven- do o uso, manutenção, limpeza e descontaminação. Em caso de equipamentos que façam registros automáti- cos de temperatura, estes devem permitir uma verificação diária da temperatura e os dados impressos ou anotados serão armaze- nados para controle.

49
Tubos de amostragem e análise
Podem ser de polipropileno ou polietileno de alta densi- dade e não devem ser reaproveitados. Deve se evitar o uso de tubos de vidro, que podem quebrar durante o armazenamento ou transporte. Ao trocar fornecedor e/ou tipo de material, rea- lizar teste de recuperação e branco para verificar se não existe interferência do material no resultado das análises.

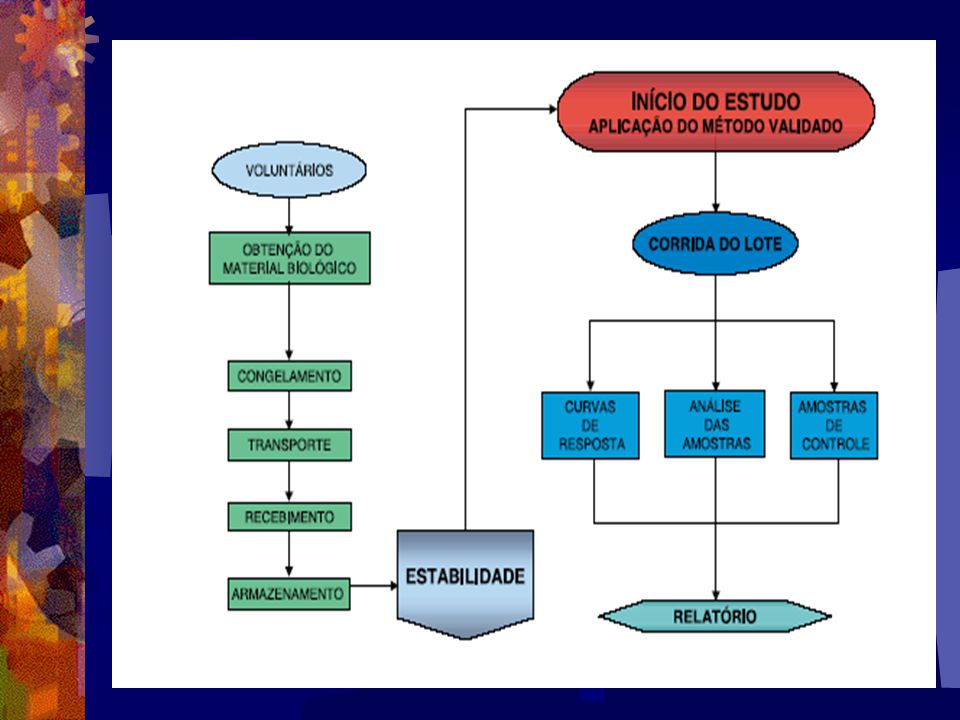
50
Protocolo do Ensaio Bioanalítico
Uma vez que o método bioanalítico foi desenvolvido e total- mente validado, um manual de procedimentos operacionais pa- dronizados (POPs) para o mesmo deve ser documentado. Portanto, todas as análises do método em questão devem ser submetidas conforme procedimentos do respectivo POP. Alterações podem afetar significativamente resultados finais como por exemplo, a substituição por um outro tipo de coluna, uma mudança significativa no comprimento de onda durante as análises e ou mudança no procedimento de extração, requer, no mínimo, revalidação parcial do método analítico.
 para o mesmo deve ser documentado. Portanto, todas as análises do método em questão devem ser. submetidas conforme procedimentos do respectivo POP. Alterações podem afetar significativamente resultados finais. como por exemplo, a substituição por um outro tipo de coluna, uma mudança significativa no comprimento de onda durante as. análises e ou mudança no procedimento de extração, requer, no. mínimo, revalidação parcial do método analítico.")
51
“A dimensão da revalidação do método depende de
fatores que podem influenciar nos resultados e, portanto, cabe ao bioanalista avaliar a melhor conduta para a sua realização (Buick et al., 1990)”.
 .")
52
As amostras biológicas dos voluntários a serem investigadas
são processadas e totalmente “randomizadas” e pode estar com- preendidas em uma única lista ou em série de listas analíticas, conforme a necessidade do fluxo de trabalho. Assim, desde que o objetivo do estudo seja comparar concentrações das amostras analíticas da droga teste em relação a droga referência, é impor- tante que a análise de amostra biológica de um voluntário esteja compreendida em uma única corrida analítica com a finalidade de minimizar efeitos da variabilidade do inter-ensaio (Dighe e Adams, 1991). Geralmente, amostras biológicas são processadas individualmente ou em duplicata, sendo esta última, a maneira mais correta durante a realização dos ensaios por cromatografia.
. Geralmente, amostras biológicas são processadas. individualmente ou em duplicata, sendo esta última, a maneira. mais correta durante a realização dos ensaios por cromatografia.")
53
amostras de padrões de calibração, amostras analíticas a serem
Normalmente, uma lista analítica é constituída por lote de amostras de padrões de calibração, amostras analíticas a serem determinadas e amostras de padrões de controle de qualidade, onde estes devem estar dispostos adequadamente em intervalos com o objetivo de monitorar o desempenho do ensaio. De cin- co a oito pontos de calibração, não incluindo amostras zero, em duplicata têm sido recomendado (Shan et al., 1992; Jackson, J.A 1994) para Estudos Biofarmacêuticos. Além disso, o in- tervalo de concentrações deve ser bem amplo, e apresente uma distribuição homogênea para as diferentes concentrações nomi- nais com intuito de caracterizar adequadamente curva de cali- bração.
 para Estudos Biofarmacêuticos. Além disso, o in- tervalo de concentrações deve ser bem amplo, e apresente uma. distribuição homogênea para as diferentes concentrações nomi- nais com intuito de caracterizar adequadamente curva de cali- bração.")
54
Os critérios de aceitabilidade de lista analítica tendo em
base resultados obtidos a partir da curva de calibração e amostras dos diferentes controles de qualidade, devem ser previamente estabelecidos. Sugere-se que quatro dos seis pontos da curva de calibração estejam em torno de 20% em relação aos seus valores de concentração nominal, e que dois pontos não satisfatórios tenham concentrações diferentes de suas concentrações reais para que a lista seja considerada aceitável (Buick et al., 1990). Entretanto, têm sido propostos outros critérios de aceitabilidade que empregam diferentes tratamentos estatísticos para a precisão e exatidão como por exemplo, realização de one-way ANOVA (Karnes et al., 1991; Causey et al,. 1990).
. Entretanto, têm sido propostos outros critérios de aceitabilidade que empregam diferentes tratamentos estatísticos para a precisão e exatidão como por exemplo, realização de one-way ANOVA (Karnes et al., 1991; Causey et al,. 1990).")
55
Parâmetros da Validação Bioanalítica
Especificidade/Seletividade Os termos especificidade e seletividade são muitas vezes permutáveis quando empregados. A especificidade de um método é definida quando o mesmo produz uma única resposta analítica para um único analito. Já a seletividade é definida como a habilidade de uma técnica analítica em distinguir e quantificar, ou não, uma droga em relação a outras resposta de diferentes componentes presen- tes nos fluidos biológicos.

56
Assim, uma vez que as técnicas cromatográficas produ-
zam respostas que possam ser distinguidas das demais resultan- tes dos diferentes interferentes presentes na matriz biológica; o termo seletivo pode ser amplamente empregado como parâme- tro, na avaliação da validação dos métodos bioanalíticos (Karnes et al., 1991; Dadgar e Brunett, 1995). Diversos interferentes podem surgir durante o desen- volvimento de um método analítico, sendo de duas origens dis- tintas: Endógenos: compreende um amplo número de compostos orgânicos de alto e baixo peso molecular (hormônios, lipídeos, proteínas, etc.), metabólitos de fármacos, precursores, produtos de degradação do analito, co-administração de fármacos e vita- minas.
. Diversos interferentes podem surgir durante o desen- volvimento de um método analítico, sendo de duas origens dis- tintas: Endógenos: compreende um amplo número de compostos. orgânicos de alto e baixo peso molecular (hormônios, lipídeos, proteínas, etc.), metabólitos de fármacos, precursores, produtos. de degradação do analito, co-administração de fármacos e vita- minas.")
57
Exógenos: impurezas presentes nos reagentes empregados
no processamento das amostras analíticas, componentes utiliza- dos na manufatura dos recipientes de acondicionamento e ou uma incompleta lavagem dos mesmos (Buick et al., 1990; Dadgar et al., 1995). Para a avaliação do teste de especificidade/seletividade, recomenda-se a utilização de no mínimo seis fontes de amostras de matriz biológica branco obtida sob controladas condições em relação ao tempo de coleta, ingestão de alimentos e outros fatores intrínsecos presentes nos protocolos dos Estudos Bio- farmacêuticos. Contudo, não deverá apresentar nenhuma inter- ferência significativa nos tempos de retenção tanto do analito quanto do seu padrão interno (Shan et al., 1992; ISO/DIS 5725- , 1994).
. Para a avaliação do teste de especificidade/seletividade, recomenda-se a utilização de no mínimo seis fontes de amostras. de matriz biológica branco obtida sob controladas condições. em relação ao tempo de coleta, ingestão de alimentos e outros. fatores intrínsecos presentes nos protocolos dos Estudos Bio- farmacêuticos. Contudo, não deverá apresentar nenhuma inter- ferência significativa nos tempos de retenção tanto do analito. quanto do seu padrão interno (Shan et al., 1992; ISO/DIS , 1994).")
58
Critérios de aceitabilidade para o teste de interferência
são amplamente empregados em análises cromatográficas. Es- tudos sugerem que qualquer interferente deverá possuir um pi- co ou uma área cromatografada menor que 20% em relação à concentração nominal do LOQ (Causey et al., 1995; Doing et al., 1990 e Dadgar et al., 1995).
.")
59
Linearidade O relatório final da International Conference of Har- monization – ICH (Guideline for Validation of Analytical Pro- cedure Methodology, 1996) determina que a linearidade de um procedimento analítico é considerada como a habilidade de se obter resultados diretamente proporcionais à concentração do analito na amostra (Comission of European Communities – Comittee for Proprietary Medicinal Products 111/5626/94 final draft, 1994; Buick et al., 1990).
 determina que a linearidade de um. procedimento analítico é considerada como a habilidade de se. obter resultados diretamente proporcionais à concentração do. analito na amostra (Comission of European Communities – Comittee for Proprietary Medicinal Products 111/5626/94 final. draft, 1994; Buick et al., 1990).")
60
A avaliação da curva de calibração, requer o emprego de no
mínimo quatro níveis de concentrações nominais, porém, estu- dos (Massart et al., 1998) recomendam o uso de 5 a 8 determina- ções que correspondem ao intervalo entre o valor superior e in- ferior da substância em exame, que atendam aos requisitos de precisão, exatidão e linearidade (Pennickx et al., 1996; Shan et al., 1992; Hill et al., 1992; Metha, 1898 e Hartmann et al., 1998). Quatro fatores devem ser empregados no desenvolvimento da curva de calibração:
 recomendam o uso de 5 a 8 determina- ções que correspondem ao intervalo entre o valor superior e in- ferior da substância em exame, que atendam aos requisitos de. precisão, exatidão e linearidade (Pennickx et al., 1996; Shan et al., 1992; Hill et al., 1992; Metha, 1898 e Hartmann et al., 1998). Quatro fatores devem ser empregados no desenvolvimento. da curva de calibração:")
61
Precisão e exatidão menor ou igual a 15% nas determinações
nominais da curva de calibração, exceto para as concentrações do LOQ Precisão e exatidão menor ou igual a 20% nas determinações nominais do LOQ Pelo menos quatro das seis concentrações nominais devem estar de acordo com critérios acima mencionados, incluindo os pontos do LOQ e os pontos de calibração de menor concentra- ção Valor de coeficiente de correlação linear ou igual ou maior que 0.95

62
cia entre os resultados de análises individuais, quando o proce-
Precisão É definida como o grau de repetibilidade ou de concordân- cia entre os resultados de análises individuais, quando o proce- dimento analítico é aplicado em diversas vezes em uma mesma amostra analítica, em idênticas condições experimentais (Swartz e Krull 1998; Buick et al., 1990; Causon, R. 1997e Brittain, 1998). Além disso, é expressa como a porcentagem do coefi- ciente de variação (C.V) ou o desvio padrão relativo da média geométrica dos valores das replicatas de cada determinação nominal, a saber:
. Além disso, é expressa como a porcentagem do coefi- ciente de variação (C.V) ou o desvio padrão relativo da média. geométrica dos valores das replicatas de cada determinação. nominal, a saber:")
63
Precisão =% CV = (dp/média) x 100
dp = nc2- (c)2 : n (n-1) Onde: CV = coeficiente de variação dp = desvio padrão n = número de dados c= concentração calculada
2 : n (n-1) Onde: CV = coeficiente de variação. dp = desvio padrão. n = número de dados. c= concentração calculada.")
64
ISO (International Organization for Standardization, 1990/
De acordo com a ICH de 1996 e o relatório final da ISO (International Organization for Standardization, 1990/ 1991), a precisão pode ser medida em três diferentes níveis (Swartz e Kull ,1998; Hartmann et al., 1994): Repetibilidade (Precisão inter-ensaio): a qual avalia a pre- cisão da metodologia durante uma corrida analítica (Buick et al.,1990). Também é definida como a habilidade de repetições de uma metodologia empregada nas mesmas condições labora- toriais, por exemplo, da utilização do mesmo corpo de analistas, dos mesmos equipamentos e dos mesmos reagentes analíticos em um curto intervalo de tempo, considerando a realização do ensaio analítico em um único dia (Hartmann et al., 1994; Causon, R. 1997; Shan et al., 1992 e ISO/Dis – final draft ,1990/1991).
, a precisão pode ser medida em três diferentes níveis. (Swartz e Kull ,1998; Hartmann et al., 1994): Repetibilidade (Precisão inter-ensaio): a qual avalia a pre- cisão da metodologia durante uma corrida analítica (Buick et. al.,1990). Também é definida como a habilidade de repetições. de uma metodologia empregada nas mesmas condições labora- toriais, por exemplo, da utilização do mesmo corpo de analistas, dos mesmos equipamentos e dos mesmos reagentes analíticos. em um curto intervalo de tempo, considerando a realização do. ensaio analítico em um único dia (Hartmann et al., 1994; Causon, R. 1997; Shan et al., 1992 e ISO/Dis – final draft ,1990/1991).")
65
ensaio poderá ser determinada através de no mínimo de 5 á 10
Neste caso, autores sugerem que esta precisão intra- ensaio poderá ser determinada através de no mínimo de 5 á 10 determinações em replicata para cada nível de concentrações nominais pré-estabelecidas das amostras de controle de qualida- de. Assim, emprega-se três níveis de concentração: baixo, que pode ser o LOQ; médio e alto, que estejam compreendidos no intervalo de limite especificado do procedimento analítico (Metha, 1989; Swartz e Krull 1998; Hartmann et al., 1998 e FDA – Guideline for Industry / Bioanalytical Method Validation final draft ,1998 e 2001).
.")
66
Reprodutibilidade (Precisão inter-ensaio): demonstra o
grau de variabilidade da precisão do método analítico em dife- rentes intervalos de tempo da análise (Buick et al.,1990). Também é definida como a habilidade de repetições da mesma metodologia analítica aplicada sob diferentes condições laboratoriais como, por exemplo, mudanças de analistas, reagen- tes e equipamentos em subsequentes ocasiões que podem com- preender várias semanas ou até meses (Shan et al., 1992; USPXX 1990; Brooks, 1985; Hartmann et al, 1994 e Buick et al., 1990). Além disso, a precisão inter-ensaio é realizada de maneira similar que a precisão intra-ensaio, porém é determinada através da análises de no mínimo em 10 dias distintos (Metha, 1989), onde os resultados obtidos são interpretados através de um coe- ficiente de variação para cada nível de concentração nominal pré- estabelecidas das amostras de controle de qualidade.
. Também é definida como a habilidade de repetições da. mesma metodologia analítica aplicada sob diferentes condições. laboratoriais como, por exemplo, mudanças de analistas, reagen- tes e equipamentos em subsequentes ocasiões que podem com- preender várias semanas ou até meses (Shan et al., 1992; USPXX. 1990; Brooks, 1985; Hartmann et al, 1994 e Buick et al., 1990). Além disso, a precisão inter-ensaio é realizada de maneira. similar que a precisão intra-ensaio, porém é determinada através. da análises de no mínimo em 10 dias distintos (Metha, 1989), onde os resultados obtidos são interpretados através de um coe- ficiente de variação para cada nível de concentração nominal. pré- estabelecidas das amostras de controle de qualidade.")
67
Para a validação de metodologias bioanalíticas aplicadas em
Estudos Biofarmacêuticos, estudos determinam que os valores de aceitabilidade para a precisão intra e inter-ensaio possuem um coeficiente de variação menor ou igual a 15%, para todos os níveis de concentração das amostras de controle de qualidade, exceto para o LOQ , onde este não deve ser superior a 20% (FDA – Guideline for Industry / Bioanalytical Method Valida- tion final draft 1998 e 2001; Shan et al., 1992 e 2000 e Causon, R. 1997).
.")
68
definições tanto para precisão como para a exatidão, a ocorrên-
Organizações Internacionais, como a ISO aborda em suas definições tanto para precisão como para a exatidão, a ocorrên- cia de diferentes erros experimentais tais como: os erros “ran- domizados” ou aleatórios (conforme descrito anteriormente na definição da precisão), e erros sistemáticos. A sistematização de erros de um método analítico é a dife- rença entre o valor médio (obtido do resultado de um número (n) de diferentes experimentos) e o valor nominal aceito como referência. Portanto, a exatidão é considerada como uma defini- ção de uma constante ou um índice de erros sistemáticos deno- minada de bias (Hartmann et al., 1994).
, e erros sistemáticos. A sistematização de erros de um método analítico é a dife- rença entre o valor médio (obtido do resultado de um número. (n) de diferentes experimentos) e o valor nominal aceito como. referência. Portanto, a exatidão é considerada como uma defini- ção de uma constante ou um índice de erros sistemáticos deno- minada de bias (Hartmann et al., 1994).")
69
% BIAS = (V. experimentais- V. nominais) / V. nominais x 100
Além disso, esta também pode ser definida como o grau de concordância entre os valores individuais encontrados em rela- ção aos valores reais ou nominais (Causon, R. 1997; Swartz e Krull 1998; Buick et al., 1990; Brittain, 1998 e Mehta, 1989). A melhor maneira de reportar a exatidão de um ensaio ana- lítico é através da porcentagem do bias , calculado a saber: % BIAS = (V. experimentais- V. nominais) / V. nominais x 100
. A melhor maneira de reportar a exatidão de um ensaio ana- lítico é através da porcentagem do bias , calculado a saber: % BIAS = (V. experimentais- V. nominais) / V. nominais x 100.")
70
Contudo, esta exatidão é determinada pelas análises em re-
plicata das amostras de controle de qualidade contendo concen- trações pré-estabelecidas. Dessa forma, empregam no mínimo três níveis de concentração nominal (baixo que pode ser o LOQ médio e alto) que estejam compreendidas dentro do intervalo de calibração especificado pela metodologia analítica utilizada (Mehta, 1989; Swartz e Krull, 1998; FDA – Guideline for In- dustry / Bioanalytical Method Validation final draft, 1998 e 2001 e Shan et al., 1992 e 2000).
 que estejam compreendidas dentro do intervalo de. calibração especificado pela metodologia analítica utilizada. (Mehta, 1989; Swartz e Krull, 1998; FDA – Guideline for In- dustry / Bioanalytical Method Validation final draft, 1998 e e Shan et al., 1992 e 2000).")
71
Para a documentação da exatidão de um ensaio analítico,
autores e determinações presentes nos guias de Validação de Metodologia Bioanalítica, estabelecem o emprego de no mínimo cinco replicatas para cada concentração nominal das amostras de controle de qualidade (Shan et al.,1992 e 2000; FDA – Guideline for Industry / Bioanalytical Method Validation final draft, 1998 e 2001; Causon, R 1997 e Jackson, A. J 1994). Normalmente os ensaios para a exatidão poderão se realiza- dos em um único dia (exatidão intra-ensaio) e também em dias diferentes (exatidão inter-ensaio) (Buick et al., 1990 e Shan et al,. 1992 e 2000).
. Normalmente os ensaios para a exatidão poderão se realiza- dos em um único dia (exatidão intra-ensaio) e também em dias. diferentes (exatidão inter-ensaio) (Buick et al., 1990 e Shan et al, e 2000).")
72
Autores sugerem que os limites de aceitabilidade para exati-
dão intra e inter-ensaio devem possuir coeficiente de variação menor ou igual a 15% para todos os níveis de concentração no- minal, exceto para o LOQ, onde este não deve exceder a limite maior que 20% ( USP XXII, 1990; Brooks, 1985; Causon, R. 1997; Shan et al., 1992 e 2000 e FDA – Guideline for Industry/ Bioanalytical Method Validation final draft, 1998 e 2001).
.")
73
Sensibilidade, Limite de Detecção (LOD) e Limite de
Quantificação (LOQ) Sensibilidade e Limite de Detecção (LOD) são dois parâ- metros muito empregados na identificação da habilidade de uma técnica analítica em detectar e quantificar traços de compostos, das mais variadas propriedades químicas, em concentrações abaixo de 1mg/L em complexos químicos ou biológicos (Mehta, A.C 1989 e Miller, J.N 1982). A metodologia analítica é dito sensível quando pequenas mudanças nos padrões de calibração podem gerar grandes va- riações na resposta do instrumento analítico. Dessa maneira, conclui-se que a sensibilidade do ensaio analítico é definida pela inclinação (“slope”) da curva de calibração (IUPAC, 1976; Causon, 1997 e Mehta, 1989).
 Sensibilidade e Limite de Detecção (LOD) são dois parâ- metros muito empregados na identificação da habilidade de uma. técnica analítica em detectar e quantificar traços de compostos, das mais variadas propriedades químicas, em concentrações. abaixo de 1mg/L em complexos químicos ou biológicos. (Mehta, A.C 1989 e Miller, J.N 1982). A metodologia analítica é dito sensível quando pequenas. mudanças nos padrões de calibração podem gerar grandes va- riações na resposta do instrumento analítico. Dessa maneira, conclui-se que a sensibilidade do ensaio analítico é definida pela. inclinação ( slope ) da curva de calibração (IUPAC, 1976; Causon, 1997 e Mehta, 1989).")
74
Por outro lado, o Limite de Detecção representa a mais baixa
concentração do composto de investigação que pode ser detec- tada em relação à amostra biológica branco com certo limite de confiabilidade (Swartz e Krull, 1998; Metha, 1989; Mehta, A.C 1987; Mehta A.C 1989; Brittain, 1998 e BuicK et al.,1990). De acordo com a International Union of Pure and Applied Chemistry - IUPAC, o Limite de Detecção é estabelecido atra- vés de análises de soluções com concentrações conhecidas e de- crescentes do analito até que o nível detectável seja 2 a 3 vezes superior ao ruído da linha de base cromatográfica (IUPAC, 1978; Rutledge e Garrick, 1989 e Bressolle , Bres J. e Moulin ,1990).
. De acordo com a International Union of Pure and Applied. Chemistry - IUPAC, o Limite de Detecção é estabelecido atra- vés de análises de soluções com concentrações conhecidas e de- crescentes do analito até que o nível detectável seja 2 a 3 vezes. superior ao ruído da linha de base cromatográfica (IUPAC, 1978; Rutledge e Garrick, 1989 e Bressolle , Bres J. e Moulin. ,1990).")
75
O Limite de Quantificação representa a mais baixa concen-
tração de um composto de investigação que pode ser mensura- do de acordo com os critérios de aceitabilidade para precisão e exatidão através de um ensaio analítico (Swartz e Krull, 1998; Causon, R e Buick et al., 1990). Na avaliação da concentração do LOQ, pode-se utilizar a ra- zão 5:1 entre o sinal e o ruído da linha de base cromatográfica no tempo de retenção do analito, ou seja, deverá obter uma tí- pica resposta do instrumento que seja cinco vezes maior que qualquer interferência na amostra branco no tempo de retenção do composto de interesse.
. Na avaliação da concentração do LOQ, pode-se utilizar a ra- zão 5:1 entre o sinal e o ruído da linha de base cromatográfica. no tempo de retenção do analito, ou seja, deverá obter uma tí- pica resposta do instrumento que seja cinco vezes maior que. qualquer interferência na amostra branco no tempo de retenção. do composto de interesse.")
76
Para a determinação do LOQ, normalmente é utilizado uma
série de análises de amostras analíticas em replicata consideran- do de 5 a 6 determinações para cada nível de concentração pré- estabelecida das amostras de controle de qualidade do LOQ (Shan et al.,1992 e 2000; Jackson, A.J 1994 e Causon, R.1997). Os critérios de aceitabilidade para a precisão e a exatidão do LOQ recomendam que o coeficiente de variação não seja maior que 20% em relação à concentração nominal (Shan et al., 1992 e 2000; Jackson, A.J 1994 e Dadgar et al., 1995).
. Os critérios de aceitabilidade para a precisão e a exatidão do. LOQ recomendam que o coeficiente de variação não seja maior. que 20% em relação à concentração nominal (Shan et al., 1992 e. 2000; Jackson, A.J 1994 e Dadgar et al., 1995).")
77
mente aplicados em diferentes etapas do ensaio da metodologia
Recuperação Os testes para a avaliação da recuperação são particular- mente aplicados em diferentes etapas do ensaio da metodologia analítica. Porém, são normalmente realizados durante a fase inicial do desenvolvimento analítico para determinar uma me- lhora da eficiência dos procedimentos de extração nas amostras biológicas, além disso, podem ser considerados como um indi- cativo da robustez do método analítico (Mehta, 1989 e Buick et al., 1990). Contudo, têm por finalidade verificar possíveis perdas durante o tratamento das amostras biológicas, como por exem- plo, da utilização de um líquido extrator (Mehta, 1989).
. Contudo, têm por finalidade verificar possíveis perdas. durante o tratamento das amostras biológicas, como por exem- plo, da utilização de um líquido extrator (Mehta, 1989).")
78
A recuperação pode ser definida como uma medida percen-
tual da eficiência dos procedimentos extrativos de um método analítico dentro de um limite de variação de aceitabilidade (FDA – Guideline for Industry / Bioanalytical Method Validation final draft,1998 e 2001). Normalmente, os experimentos para a avaliação da recupera- ção são realizados através da contaminação de três níveis (baixo, médio e alto) de concentração nominal do composto analítico nas amostras biológicas branco. Sugere-se no mínimo o empre- go de cinco replicatas para cada concentração, onde estas deve- rão estar compreendidas em intervalo de calibração especificado pela metodologia analítica (Mehta, 1989; Causon, R e FDA – Guideline for Industry / Bioanalytical Method Valida- tion final draft ,1998 e 2001).
. Normalmente, os experimentos para a avaliação da recupera- ção são realizados através da contaminação de três níveis (baixo, médio e alto) de concentração nominal do composto analítico. nas amostras biológicas branco. Sugere-se no mínimo o empre- go de cinco replicatas para cada concentração, onde estas deve- rão estar compreendidas em intervalo de calibração especificado. pela metodologia analítica (Mehta, 1989; Causon, R e. FDA – Guideline for Industry / Bioanalytical Method Valida- tion final draft ,1998 e 2001).")
79
Existem várias formas utilizadas na determinação da recupe-
ração do ensaio analítico. A maneira mais usual para a quantifi- cação da recuperação é através da comparação da resposta da resultante do analito (pico ou área cromatografada) das amos- tras analíticas em replicata, previamente tratadas em relação as mesmas em idênticas concentrações preparadas em fase móvel, correspondendo 100% da recuperação (Jackson, J.A 1994; Taylor, J.K 1987; Causon, R. 1997; Buick et al., 1990; Mehta, 1989; Brooks, 1987 e Dadgar et al., 1995). A recuperação de um analito não precisa ser 100%, mas a quantidade de analito recuperado e do padrão interno deve ser consistente, precisa e reprodutível, ou seja ter valores dentro do limite de aceitabilidade para precisão e exatidão..
 das amos- tras analíticas em replicata, previamente tratadas em relação as. mesmas em idênticas concentrações preparadas em fase móvel, correspondendo 100% da recuperação (Jackson, J.A 1994; Taylor, J.K 1987; Causon, R. 1997; Buick et al., 1990; Mehta, 1989; Brooks, 1987 e Dadgar et al., 1995). A recuperação de um analito não precisa ser 100%, mas a. quantidade de analito recuperado e do padrão interno deve ser. consistente, precisa e reprodutível, ou seja ter valores dentro. do limite de aceitabilidade para precisão e exatidão..")
80
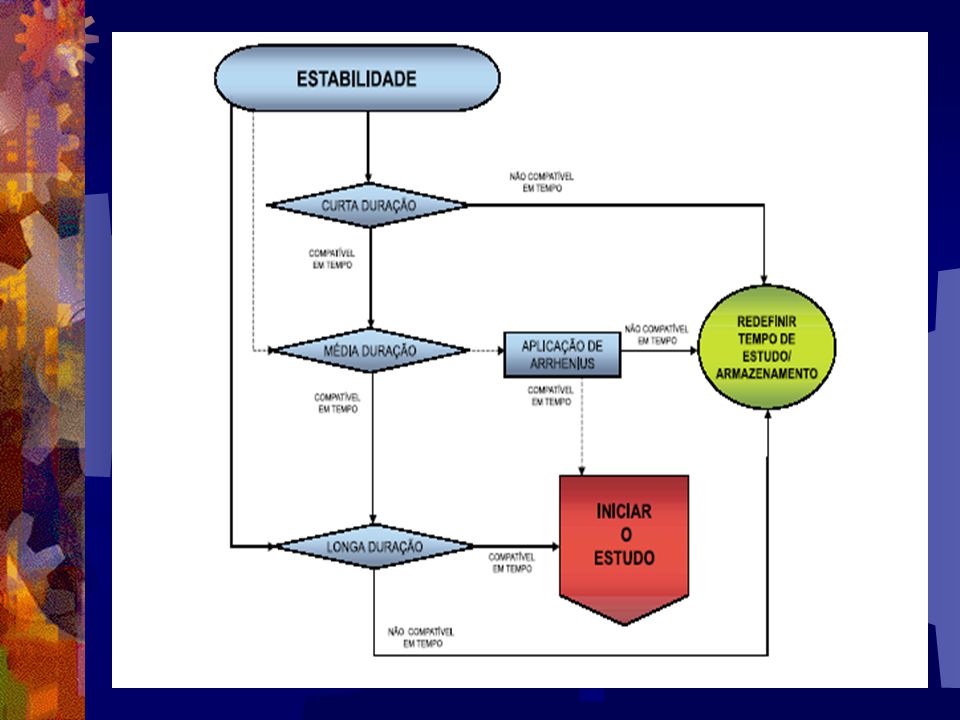
Estabilidade A Resolução . RDC Nº 84 . de 19 de março de 2002, estabeleceu quais os tipos de estabilidade em fluidos biológicos que devem ser realizados na fase pré-estudo de bioequivalência visando a validação do método para fins do estudo propriamen- te dito. Neste sentido, os estudos de estabilidade foram classifi- cados em: curta duração; média duração; longa duração; de soluções-padrão;

81
Estabilidade de curta duração
Congelamento e descongelamento A estabilidade do analito deve ser determinada após 3 ci- clos de congelamento e descongelamento.No mínimo 3 alíquo- tas a cada concentração (alta e baixa) devendo ser estocado a cada temperatura pretendida por 24 horas e descongelada sem auxílio a temperatura ambiente. Quando completamente des- congelada, as amostras devem ser re-congeladas por 12 a 24 horas sob as mesmas condições. Os ciclos de congelamento e descongelamento devem ser repetidos por 3 vezes e analisados no terceiro ciclo. Se um fármaco é instável à temperatura ambiente, por exemplo, as amostras de estabilidade devem ser congeladas a . 20 ou . 70ºC durante 3 ciclos de congelamento e descongelamento.
 devendo ser estocado a. cada temperatura pretendida por 24 horas e descongelada sem. auxílio a temperatura ambiente. Quando completamente des- congelada, as amostras devem ser re-congeladas por 12 a 24. horas sob as mesmas condições. Os ciclos de congelamento e. descongelamento devem ser repetidos por 3 vezes e analisados. no terceiro ciclo. Se um fármaco é instável à temperatura. ambiente, por exemplo, as amostras de estabilidade devem ser. congeladas a . 20 ou . 70ºC durante 3 ciclos de congelamento e. descongelamento.")
82
Condições de análise Três (03) alíquotas de cada concentração (alta e baixa) devem ser descongeladas a temperatura ambiente e deixadas nesta tem- peratura durante o tempo máximo da análise do lote. A estabi- lidade das amostras processadas, incluindo o tempo de residên- cia no auto-injetor, deve ser determinada. Deve ser demonstra- do que os fármacos permanecem intactos se deixados por várias horas à temperatura ambiente na matriz biológica. Certos fár- macos, por exemplo, captopril, AAS, etc., sofrem mudanças imediatas, degradação em matrizes biológicas. Em tais casos, aditivos apropriados e/ou agentes derivatizantes podem ser adicionados.

83
Estabilidade de média duração
A estabilidade de média duração deve ser determinada pelo armazenamento de no mínimo cinco alíquotas de cada concentração ( alta, média e baixa) na temperatura de -20°C . As amostras para este estudo devem ser analisadas nos tempo zero, um, dois quatro, oito e dezesseis dias de armazena- mento. Os dados devem ser tratados com a utilização da equa- ção de .Arrhenius. Para cálculo da constante de velocidade de reação e do tempo de armazenamento na temperatura estudada. Com os dados de estabilidade obtida e viáveis com o tempo analítico, pode-se iniciar a internação ou o estudo sobre os indivíduos.
 na temperatura de -20°C . As amostras para este estudo devem ser analisadas nos. tempo zero, um, dois quatro, oito e dezesseis dias de armazena- mento. Os dados devem ser tratados com a utilização da equa- ção de .Arrhenius. Para cálculo da constante de velocidade de. reação e do tempo de armazenamento na temperatura estudada. Com os dados de estabilidade obtida e viáveis com o. tempo analítico, pode-se iniciar a internação ou o estudo sobre. os indivíduos.")
84
Estabilidade de longa duração
O tempo de estocagem num estudo de estabilidade de longa duração deve exceder o tempo entre a data da primeira coleta das amostras e a data da análise da última amostra. A es- tabilidade de longa duração deve ser determinada pela estoca- gem de no mínimo 3 alíquotas de cada concentração (alta, mé- dia e baixa) sob as mesmas condições das amostras de estudo. Normalmente, é realizada em 03 temperaturas de estoca- gem,4, -20 e .70ºC. O volume das amostras deve ser suficiente para as análises no mínimo em 03 tempos diferentes (FDA, 1997). As concentrações médias das determinações de todas as concentrações utilizadas devem ser comparadas com as médias obtidas das análises das amostras recém-preparadas para os es- tudos de estabilidade de longa duração, ou seja, no tempo zero.
 sob as mesmas condições das amostras de estudo. Normalmente, é realizada em 03 temperaturas de estoca- gem,4, -20 e .70ºC. O volume das amostras deve ser suficiente. para as análises no mínimo em 03 tempos diferentes (FDA, 1997). As concentrações médias das determinações de todas as. concentrações utilizadas devem ser comparadas com as médias. obtidas das análises das amostras recém-preparadas para os es- tudos de estabilidade de longa duração, ou seja, no tempo zero.")
85
assegurados no tempo necessário para análise de todo o lote das
Soluções-padrão A estabilidade do fármaco e padrão interno devem ser assegurados no tempo necessário para análise de todo o lote das amostras, incluindo possíveis interrupções acidentais. Dados da literatura ou testes de laboratório devem ser conduzidos para determinar se o fármaco puro ou em mistura com metabólitos e padrão interno dissolvidos num sistema de solvente são estáveis sob as condições de ensaios, especificamente em relação aos fa- tores físicos, tais como:calor, umidade, luz e exposição ao ar. A estabilidade da solução estoque, contendo o fármaco e padrão interno, deve ser avaliada à temperatura ambiente por no mínimo 6 horas. Se a solução estoque e padrão interno são refrigeradas ou congeladas por um período relevante, a estabili- dade deve ser documentada.

86
Após completar o tempo de estocagem desejado, a estabili-
dade deve ser testada em comparação com a solução preparada recentemente.

87
Robustez A robustez é um parâmetro que refere se a reprodutibili- dade de uma metodologia analítica sob diferentes condições ex- perimentais (Shan et al., 1992; Jackson, A.J 1994; Swartz e Krull, 1998). Normalmente, a robustez pode ser avaliada experimen- talmente através de alterações das condições experimentais do ensaio analítico (Youden & Steiner, 1975 ; Swartz e Krull,1998). É desejável que um procedimento analítico seja capaz de resistir frente às alterações das condições experimentais. Cons- tatando a suscetibilidade nos parâmetros analíticos (precisão, exatidão, linearidade, etc.), estes deverão ser adequadamente controlados ou será necessário à inclusão de novas precauções na metodologia (Shan et al., 1992; Swartz e Krull, 1998 e Jack- son A.J 1994).
. Normalmente, a robustez pode ser avaliada experimen- talmente através de alterações das condições experimentais do. ensaio analítico (Youden & Steiner, 1975 ; Swartz e Krull,1998). É desejável que um procedimento analítico seja capaz de. resistir frente às alterações das condições experimentais. Cons- tatando a suscetibilidade nos parâmetros analíticos (precisão, exatidão, linearidade, etc.), estes deverão ser adequadamente. controlados ou será necessário à inclusão de novas precauções. na metodologia (Shan et al., 1992; Swartz e Krull, 1998 e Jack- son A.J 1994).")
88
Para minimizar os efeitos da variabilidade no método analí-
tico, é necessário investigar e documentar influências durante o processo de validação analítica, as quais podemos citar (Dadgar et al.,1995; Shan et al., 1992 ; Jackson, A.J 1994): Mudanças das condições da temperatura ambiente Alterações do corpo de analista Mudanças de equipamentos Tempo de extração Variações típicas em cromatografia líquida como:
: Mudanças das condições da temperatura ambiente. Alterações do corpo de analista. Mudanças de equipamentos. Tempo de extração. Variações típicas em cromatografia líquida como:")
89
Influência da variação de pH da fase móvel;
Influência da variação da composição da fase móvel; Diferentes colunas cromatográficas; Temperatura; Velocidade de fluxo

90
Portanto, a avaliação destas influências acima citadas, forne-
cerá indicativo da habilidade da metodologia de se manter está- vel frente às alterações das condições analíticas (Dadgar et al., 1995). De acordo com a ICH, a robustez deve ser considerada na fase de desenvolvimento e será convenientemente monitorada durante o ensaio analítico, observando possíveis mudanças na linearidade e nos resultados das amostras de controle de quali- dade (Jackson, A.J 1994; Dadgar et al., 1995; Swartz e Krull, 1998 e Shan et al., 1992).
. De acordo com a ICH, a robustez deve ser considerada na. fase de desenvolvimento e será convenientemente monitorada. durante o ensaio analítico, observando possíveis mudanças na. linearidade e nos resultados das amostras de controle de quali- dade (Jackson, A.J 1994; Dadgar et al., 1995; Swartz e Krull, 1998 e Shan et al., 1992).")
Apresentações semelhantes