Carregar apresentação
A apresentação está carregando. Por favor, espere

1
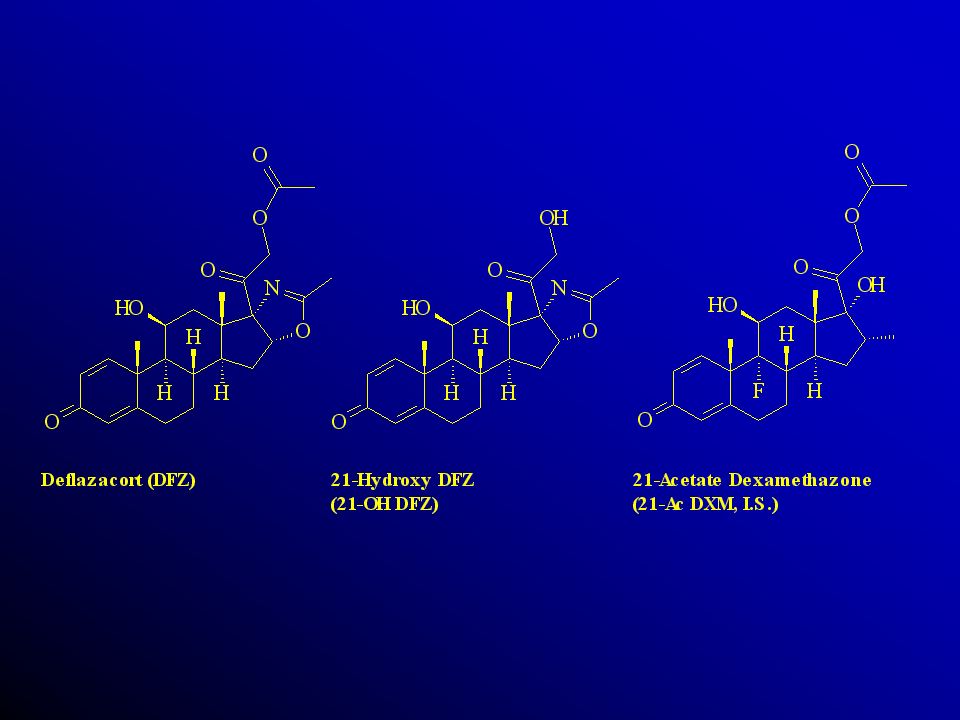
Validação de Métodos Bioanalíticos Curso de Farmacologia Clínica
Universidade Mogi das Cruzes Abril 2004

2
Nas últimas décadas, houve um aumento de
métodos bioanalíticos aplicados à cromatografia para a determinação qualitativa e quantitativa de fármacos, produtos acabados, matérias primas e amostras biológicas, em todas as fases do desen- volvimento do fármaco desde a pesquisa até a rea- lização de estudos de bioequivalência.

3
Assim, devido este aumento e a globalização
da economia, a adoção de um sistema de qualida- de reconhecido universalmente aceito, é requisito fundamental.

4
Portanto, o entendimento do que constituí
uma validação de métodos cromatográficos, possi- bilita a garantia de um nível satisfatório de qualida- de na validação metodológica (Massart et al., 1998; Swartz & Krull, 1998).
.")
5
Desde o final dos anos 80, a importância da
validação de métodos bioanalíticos e suas influên- cias na avaliação e interpretação dos resultados, transformou-se em alvo de amplas discussões pre- sentes em diversas Conferências tanto nos EUA quanto na Europa e consequentemente fornecer adequadas diretrizes em relação aos parâmetros exigidos na validação analítica (Shan et al., 1991 e 1992; Dadgar, 1995; Cartwright et al., 1991 e Arnoux & Morrison, 1992) .
 .")
6
A correta avaliação dos Estudos Biofarma-
cêuticos só pode ser alcançada se a exatidão dos dados analíticos forem obtidos (Pachla et al.,1986). Além disso, essa exatidão depende de crité- rios fundamentais empregados, não só em relação à adequada interpretação desses resultados, como também na aplicação da confiabilidade e na totali- dade do desempenho do método bioanalitico.
. Além disso, essa exatidão depende de crité- rios fundamentais empregados, não só em relação. à adequada interpretação desses resultados, como. também na aplicação da confiabilidade e na totali- dade do desempenho do método bioanalitico.")
7
Assim, podemos destacar os principais critérios
fundamentais empregados na validação de métodos bioanalíticos:

8
Avaliação da droga e estabilidade do analito .
Seletividade/Especificidade. Linearidade e modelo de calibração. Precisão e Exatidão. Sensibilidade, Limite de Quantificação e Detecção Recuperação * Pachla et al., 1986; Buick et al., 1990; MacDouglas e Crummett, 1980; Taylor, 1983; Brooks e Weinfeld,1985; Dadgar e Smith, 1986; Inman et al., 1987 e Shan et al., 1987.

9
Modernos métodos bioanalíticos são funda-
mentados em relação a uma variedade de técnicas físicoquímicas e biológicas á saber:

10
Métodos Químicos: Cromatografia (CG, HPLC) e uma variedade de processos utilizando métodos de espectrometria de massa (MS) tais como MS-MS e combinações de técnicas tais como CG-MS,LC-MS. Métodos Biológicos: baseados em procedimen- tos de imunoensaios; em particular o Radioimu-noensaio (RIA), imunoensaio de múltiplas enzi-mas (EMIT) e ensaio de imunoabsorção de enzi-mas ligadas (ELISA). Métodos microbiológicos
, imunoensaio de múltiplas enzi-mas (EMIT) e ensaio de imunoabsorção de enzi-mas ligadas (ELISA). Métodos microbiológicos.")
13
Tais métodos analíticos devem ser cuidado-
samente pré- validados na fase de desenvolvimento e também validados durante o ensaio analítico, para garantir que os mesmos sejam satisfatoriamente realizados e que a confirmação das especificações pré-determinadas seja encontrada; além disso, gerar confiabilidade nos resultados obtidos (Buick et al., 1990; Jackson, J. A; 1994; Shan et al., 1992).
.")
14
Para tanto, os mesmos envolvem procedimen-
tos de determinação e quantificação de moléculas orgânicas com amplas propriedades físico- quími- cas, onde cada composto deve ser avaliado de acor- do com as suas propriedades individuais e também levando em consideração a complexidade analítica e as concentrações almejadas (Jackson, J. A 1994).
.")
15
Seleção e Desenvolvimento
Pode-se considerar que a etapa crítica no Es- tudo Biofarmacêutico é o desenvolvimento e valida- ção de ensaios bio-analíticos, que consistem de experimentos realizados para encontrar condições necessárias para a quantificação do analito em questão.

16
Isto requer conhecimentos das propriedades
físico-químicas da droga para que o processo de desenvolvimento seja racional e individualizado. Assim, todos os critérios envolvidos no desen- volvimento analítico deverão ser registrados em relatórios de acompanhamento analítico (Snyder, 1988; Jackson, J.A 1994).
.")
17
Padrões Análise de Drogas ou seus Metabólitos é invariávelmete feita através de amostras “contaminadas”, curvas de calibração e amostras de controle de qualidade (QC). Padrões de Referência autenticados devem ser utilizados no estudo. Preferivelmente o mesmo composto. Caso contrário um sal, base, ácido ou éster do composto pode ser usado.
. Padrões de Referência autenticados devem ser utilizados no estudo. Preferivelmente o mesmo composto. Caso contrário um sal, base, ácido ou éster do composto pode ser usado.")
18
Escolha da Procedência:
Padrões certificados (U.S.Pharmacopea) Padrões disponíveis comercialmente de fontes de conhecida reputação. Padrões com pureza documentada, específicamente sintetízados por laboratórios comerciais.
 Padrões disponíveis comercialmente de fontes de conhecida reputação. Padrões com pureza documentada, específicamente sintetízados por laboratórios comerciais.")
20
A realização de uma pesquisa bibliográfica é
a primeira etapa para a busca do método bioanalíti- co. Uma vez existindo o método, ele deverá ser tes- tado quanto a sua reprodutibilidade. Na inexistênciade um método bioanalítico para um determinado fármaco, o centro analítico deve desenvolver um método que responda satisfatória- mente ao estudo desejado..

21
Para a aplicação deste fato, consideram-se os
seguintes tipos de validação: Validação Total Validação total é de importância no desen- volvimento e implementação de um método quando aplicado pela primeira vez ou quando for utilizado para determinar um novo analito nesta mesma condição analítica.

22
Validações parciais são modificações do
Validação Parcial Validações parciais são modificações do método já validado. Uma validação parcial pode compreender desde uma pequena determinação de precisão/exatidão a até quase uma validação total.

23
No desenvolvimento de um método é nece-
ssário verificar toda a metodologia de preparação da amostra, a qual envolve os processos de extra- ção, separação, purificação, identificação e quanti- ficação do fármaco na matriz biológica. Para tanto, podemos dizer que nesta fase, temos duas etapas distintas á saber:

24
Pré-Validação Objetivo: Avaliar os parâmetros de desempenho do método. Deve ser realizado para cada matriz biológica e cada espécie química estudada. Deve ser realizado com pelo menos 3 lotes da matriz biológica, onde cada lote é coletado de uma fonte diferente.

25
Cada lote deve conter: Uma curva de calibração utilizando a matriz pura, matriz + I.S. e 5 a 8 pontos com a faixa de aplicação do método. Amostras de Controle (n 5) de Qualidade de Baixa concentração (QCA) Média concentração e (QCB) Alta concentração (QCC) e Limite de Detecção (LOQ QC).
 de Qualidade de Baixa concentração (QCA) Média concentração e (QCB) Alta concentração (QCC) e Limite de Detecção (LOQ QC).")
26
Nesta fase consiste na realização de alguns
ensaios preliminares visando à determinação dos seguintes parâmetros: Exatidão, precisão e recuperação; Linearidade e limites de quantificação; Seletividade.

27
Validação do Método É a fase propriamente dita do desenvolvimen- to e estabelecimento da metodologia com o objeti- vo de definir o ensaio bioanalítico. Neste caso, a padronização de um método bio-analítico típico inclui a determinação dos se- guintes parâmetros fundamentais á saber:

28
Especificidade e Seletividade;
Linearidade; Precisão e Exatidão; Sensibilidade, Limite de quantificação e detecção; Recuperação; Estabilidade ;

29
Protocolo do Ensaio Bioanalítico
Uma vez que o método bioanalítico foi desen- volvido e totalmente validado, um manual de proce- dimentos operacionais padronizados (POPs) para o mesmo deve ser documentado. Portanto, todas as análises do método em questão devem ser submetidas conforme procedi- mentos do respectivo POP.
 para o. mesmo deve ser documentado. Portanto, todas as análises do método em. questão devem ser submetidas conforme procedi- mentos do respectivo POP.")
30
Alterações podem afetar significativamente
resultados finais como por exemplo, a substituição por um outro tipo de coluna, uma mudança signifi- cativa no comprimento de onda durante as análises e ou mudança no procedimento de extração, requer, no mínimo, revalidação parcial do método analítico.

31
As amostras biológicas dos voluntários a
serem investigadas são processadas preferencial- mente em duplicata e totalmente “randomizadas” e podem estar compreendidas em: Uma única lista Em série de listas analíticas

32
Importante que a análise de amostra bioló-
gica de um voluntário esteja compreendida em uma única corrida analítica com a finalidadede minimizar efeitos da variabilidade do inter-ensaio (Dighe e Adams, 1991).
.")
33
Uma Lista analítica é constituída por * :
Amostras analíticas em duplicata a ser determinadas. Uma curva de calibração em duplicata utilizando a matriz pura, matriz + I.S. e 5 a 8 pontos correspondendo faixa de aplicação do método. * Shan et al., 1992; Jackson, J.A 1994

34
Amostras de Controle (n 5) de Qualidade de Baixa concentração (QCA) Média concentração e (QCB) Alta concentração (QCC) dispostos em intervalos para monitorar o desempenho analítico.
 de Qualidade de Baixa concentração (QCA) Média concentração e (QCB) Alta concentração (QCC) dispostos em intervalos para monitorar o desempenho analítico.")
35
Parâmetros da Validação Bioanalítica

36
Especificidade - Seletividade
A especificidade de um método é definida quando o mesmo produz uma única resposta analí- tica para um único analito.

37
Já a seletividade é definida como a habilidade
de uma técnica analítica em distinguir e quantificar, ou não, uma droga em relação a outras resposta de diferentes componentes presentes nos fluidos bio- lógicos.

38
Assim, uma vez que as técnicas cromatográ-
ficas produzam respostas que possam ser distin- guidas das demais resultantes dos diferentes inter- ferentes presentes na matriz biológica; o termo se- letivo é usualmente aplicado como parâmetro, na validação dos métodos bioanalíticos (Karnes et al., 1991; Dadgar e Brunett, 1995).
.")
39
Diversos interferentes podem surgir durante
o desenvolvimento de um método analítico, á saber: Endógenos: compreende um amplo número de compostos orgânicos de alto e baixo peso molecular (hormônios, lipídeos, proteínas, etc.), metabólitos de fármacos, precursores, produtos de degradação do analito, co-administração de fármacos e vitaminas.
, metabólitos de fármacos, precursores, produtos. de degradação do analito, co-administração de fármacos e vitaminas.")
40
Exógenos: impurezas presentes nos reagentes empregados no processamento das amostras analíticas, componentes utilizados na manufatura dos recipientes de acondicionamento e ou uma incompleta lavagem dos mesmos (Buick et al., 1990; Dadgar et al., 1995).
.")
41
Análise da especificidade e seletividade emprega-sede no mínimo seis fontes de amostras de matriz biológica branco obtida sob controladas condições, avaliando-se o horário da coleta, ingestão de alimentos, e outros fatores considerados importantes ao método. Cada “branco” deve ser avaliado em relação a interferentes, utilizando-se o método de extração proposto e as condições analíticas em questão (HPLC, LC/MS/MS).
.")
42
Os resultados são confrontados com os obtidos de uma solução aquosa do analíto em concentrações ao Limite de Quantificação (LOQ). Qualquer interferência significativa no tempo de retenção da droga, do metabólitos ou do padrão interno (I.S.) deve ser rejeitado (Shan et al., 1992; ISO/DIS , 1994).
 deve ser rejeitado (Shan et al., 1992; ISO/DIS , 1994).")
43
Interferência em mais que 20% em relação a concentração do LOQ, o método deve ser alterado para eliminar os interferentes (Causey et al., 1995; Doing et al., 1990 e Dadgar et al., 1995). Nicotina, drogas OTC e seus metabólitos, como cafeína, aspirina, acetoaminofen e ibuprofeno devem ser testados. Se mais de um analíto é avaliado, cada um deve ser injetado separadamente.

45
Blank Sample I.S. 21-OH DFZ 21-OH DFZ (1ng/mL) + I.S. I.S. 21-OH DFZ
 + I.S. I.S. 21-OH DFZ")
46
*International Conference of Harmonization - ICH, Guideline
Linearidade* É considerada como a habilidade de se obter resultados diretamente proporcionais à concentra- ção do analito na amostra. *International Conference of Harmonization - ICH, Guideline for Validation of Analytical Procedure Methodology, 1996; Comissionof European Communities – Comittee for Proprie- tary Medicinal Products 111/5626/94 final draft, 1994; Buick et al., 1990.

47
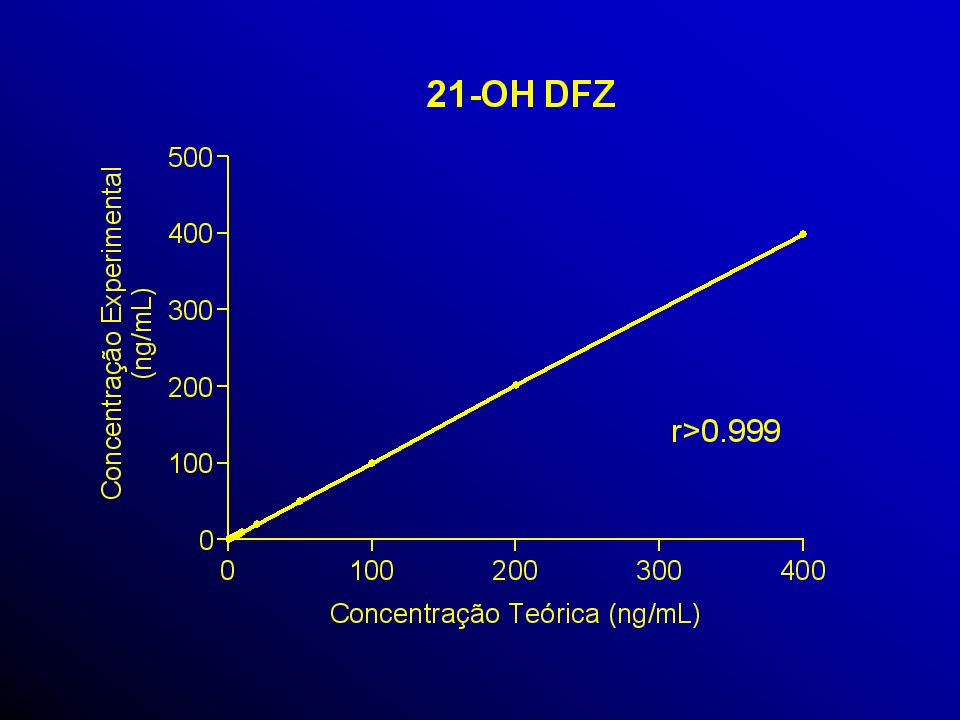
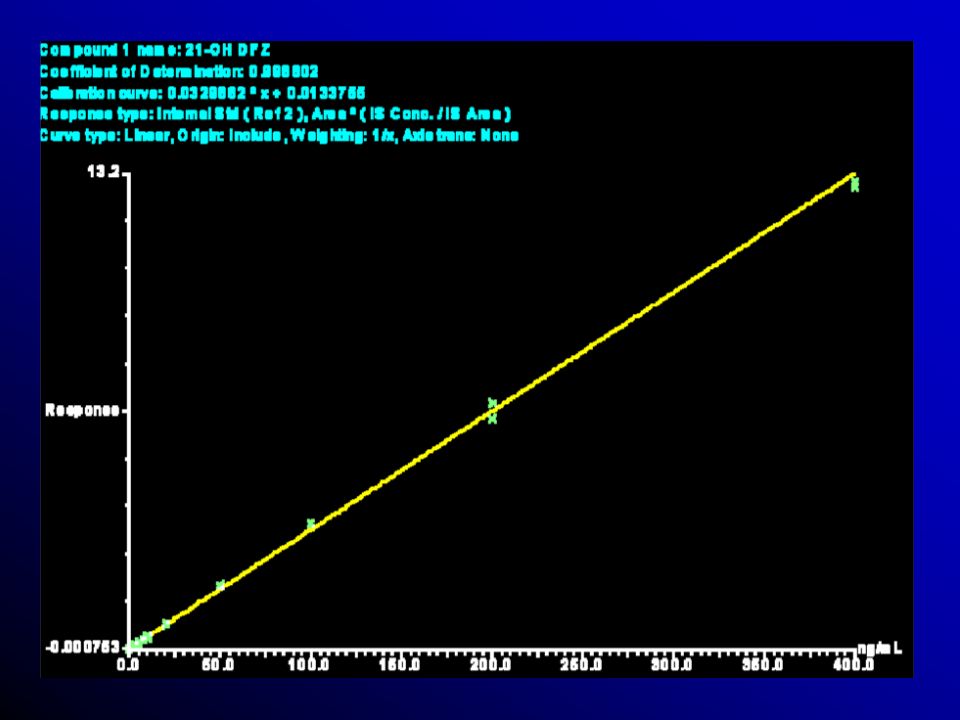
A avaliação da curva de calibração, recomen- rdam o uso:
5 a 8 determinações que correspondem ao intervalo entre o valor superior e inferior da substância em exame, que atendam aos requisitos de precisão, exatidão e linearidade. Pennickx et al., 1996; Shan et al., 1992; Hill et al., 1992; Metha, 1898; Massart et al., 1998 e Hartmann et al., 1998.

48
Quatro fatores devem ser empregados no
desenvolvimento da curva de calibração: Precisão e exatidão menor ou igual a 15% nas determinações nominais da curva de calibração, exceto para as concentrações do LOQ. Precisão e exatidão menor ou igual a 20% nas determinações nominais do LOQ.

49
Pelo menos quatro das seis concentrações nominais devem estar de acordo com critérios acima mencionados, incluindo os pontos do LOQ e os pontos de calibração de menor concentra-ção. Valor de coeficiente de correlação linear ou igual ou maior que 0.95.

51
O Padrão Interno (I.S.) tem como finalidade corrigir erros e\ou perdas
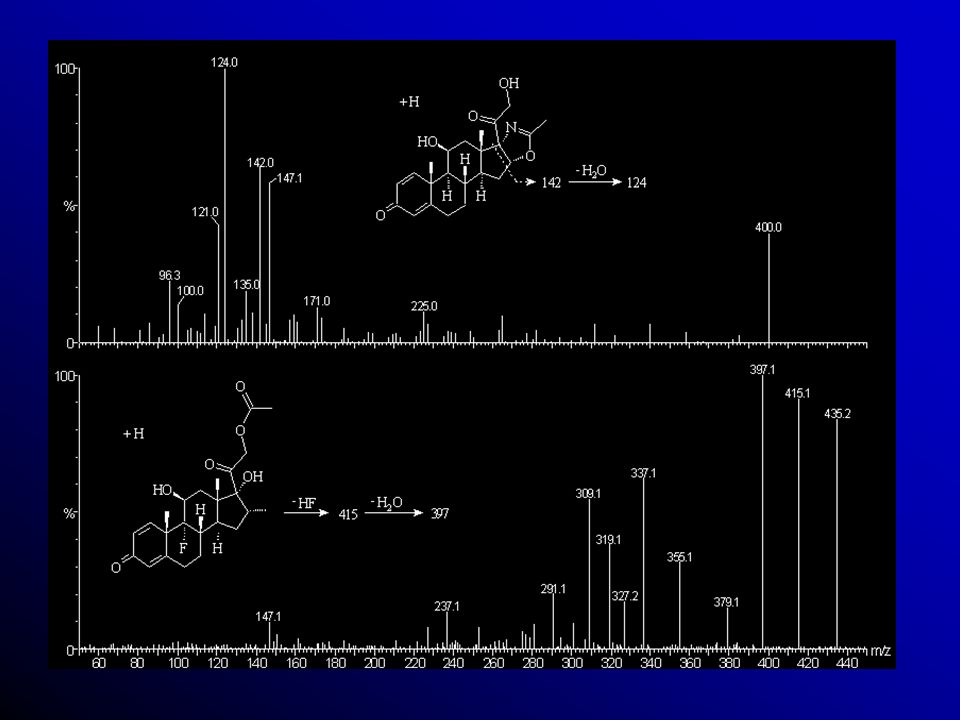
2 1 - H y d r o x D F Z ( O ) N O Padrão Interno (I.S.) tem como finalidade corrigir erros e\ou perdas por transferências e pipetagem. Uma vez que sua concentração é conhecida podemos corrigir a concentração do analito desassociando o volume. 100ng/mL 100L Razão: 5 2 1 - A c e t a D x m h z o n ( X M , I . S ) O H F 20ng/mL
 N. O Padrão Interno (I.S.) tem como finalidade corrigir erros e\ou perdas. por transferências e pipetagem. Uma vez que. sua concentração é conhecida podemos corrigir a concentração do analito desassociando o volume. 100ng/mL. 100L. Razão: A. c. e. t. a. D. x. m. h. z. o. n. ( X. M. , I. . S. ) O. H. F. 20ng/mL.")
53
Voluntário 08 - 1:30h após administração
21-OH DFZ

54
Precisão e Exatidão Exatidão Ruim Exatidão Ruim Precisão Ruim
Precisão Boa Exatidão Boa Precisão Ruim Exatidão Boa Precisão Boa

55
Precisão os resultados de análises individuais,quando o pro-
É definida como grau de concordância entre os resultados de análises individuais,quando o pro- cedimento analítico é aplicado em diversas vezes em uma mesma amostra analítica, em idênticas condições experimentais. Swartz e Krull 1998; Buick et al., 1990; Causon, R e Brittain,

56
Precisão =% CV = (dp/média) x 100
Além disso, é expressa como a porcentagem do coeficiente de variação (C.V) ou o desvio padrão relativo da média geométrica dos valores das repli- catas de cada determinação nominal : Precisão =% CV = (dp/média) x 100 dp = nc2- (c)2 : n (n-1) Onde: CV = coeficiente de variação dp = desvio padrão n = número de dados c= concentração calculada
 ou o desvio padrão. relativo da média geométrica dos valores das repli- catas de cada determinação nominal : Precisão =% CV = (dp/média) x 100. dp = nc2- (c)2 : n (n-1) Onde: CV = coeficiente de variação. dp = desvio padrão. n = número de dados. c= concentração calculada.")
57
* De acordo com a ICH de 1996 a precisão
pode ser medida em dois diferentes níveis : Repetibilidade (Precisão intra-ensaio): é definida como a habilidade de repetições de uma metodologia empregada nas mesmas condições laboratoriais, da utilização do mesmo corpo de analistas, dos mesmos equipamentos e dos mesmos reagentes analíticos em um curto intervalo de tempo, considerando a realização do ensaio analítico em um único dia . * Relatório final da ISO (International Organization for Standardiza- tion, 1990/1991), Swartz e Kull ,1998; Hartmann et al., 1994; Causon, R. 1997; Shan et al., 1992; Buick et al.,1990
: é definida como a habilidade de repetições de uma metodologia empregada nas mesmas condições laboratoriais, da utilização do mesmo corpo de analistas, dos mesmos equipamentos e dos mesmos reagentes analíticos em um curto intervalo de tempo, considerando a realização do ensaio analítico em um único dia . * Relatório final da ISO (International Organization for Standardiza- tion, 1990/1991), Swartz e Kull ,1998; Hartmann et al., 1994; Causon, R. 1997; Shan et al., 1992; Buick et al.,1990.")
58
Reprodutibilidade (Precisão inter-ensaio): é definida como a habilidade de repetições da mesma metodologia analítica aplicada sob diferentes condições laboratoriais como, por exemplo, mudanças de analistas, reagentes e equipamentos em subsequentes ocasiões que podem compreender várias semanas ou até meses. * Shan et al., 1992; USPXX 1990; Brooks, 1985; Hartmann et al, 1994 e Buick et al., 1990.

59
A avaliação da precisão intra e inter ensaio,
recomendam o uso: 5 á 10 determinações em replicata para cada nível de concentrações nominais pré-estabelecidas das amostras de controle de qualidade. Emprega-se três níveis de concentração: baixo, que pode ser o LOQ; médio e alto, que estejam compreendidos dentro do intervalo de limite especificado do procedimento analítico.

60
Exatidão É definida como o grau de concordância
entre os valores individuais encontrados em rela- ção aos valores reais ou nominais * Causon, R. 1997; Swartz e Krull 1998; Buick et al., 1990; Brittain, 1998 e Mehta, 1989.

61
Além disso, é expressa como a porcentagem
do erro padrão relativo da média geométrica dos valores das replicatas de cada determinação nomi- nal : % E.R = (V. exp - V. t) / V. t x 100 Onde, V. exp: Valores Experimentais V. t: Valores teóricos
 / V. t x 100. Onde, V. exp: Valores Experimentais. V. t: Valores teóricos.")
62
Normalmente os ensaios para a exatidão poderão se realizados:
Um único dia “ Exatidão Intra-ensaio” Dias diferentes “Exatidão Inter-ensaio” Emprega-se três níveis de concentração: baixo, que pode ser o LOQ; médio e alto, que estejam compreendidos dentro do intervalo de limite especificado do procedimento analítico. 5 á 10 determinações em replicata para cada nível de concentrações nominais pré-estabelecidas das amostras de controle de qualidade. * Buick et al., 1990 e Shan et al, e 2000.

63
Sensibilidade, Limite de Detecção (LOD), Limite de Quantificação (LOQ)
A metodologia analítica é dito sensível quan- do pequenas mudanças nos padrões de calibração podem gerar grandes variações na resposta do ins- trumento analítico. Portanto, ela está relacionada com inclinação (“slope”) da curva de calibração. * IUPAC, 1976;Causon, 1997 e Mehta, 1989
 da curva de calibração. * IUPAC, 1976;Causon, 1997 e Mehta,")
64
Limite de Detecção representa a mais baixa
concentração do composto de investigação que pode ser detectada em relação à amostra biológica branco com certo limite de confiabilidade. É estabelecido através de análises de soluções com concentrações conhecidas e decrescentes do analito até que o nível detectável seja 2 a 3 vezes superior ao ruído da linha de base cromatográfica * Swartz e Krull, 1998; Metha, 1989; Mehta, A.C 1987; Mehta A.C 1989; Brittain, 1998 e BuicK et al.,1990.

65
O Limite de Quantificação representa a mais
baixa concentração de um composto de investiga- ção que atende os requisitos para precisão e exati- dão do ensaio analítico. É estabelecido através da a razão 5:1 entre o sinal e o ruído da linha de base cromatográfica no tempo de retenção do analito. de 5 a 6 determinações em replicata para cada nível de concentração pré-estabelecida das amostras de controle de qualidade do LOQ. * Swartz e Krull, 1998; Causon, R e Buick et al., 1990.

66
Recuperação Recuperação de um analíto em um estudo é a
resposta obtida de uma quantidade do analito adi- cionada e recuperada da matrixbiológica e compa- rada a resposta obtida do padrão puro. A recuperação avalia a eficiência da extração e sua variabilidade. Embora recuperações próximas de 100% sejam desejáveis, baixas recuperações po- dem ser utilizadas (50-60%) se forem precisas, exa- tas e reprodutíveis.
 se forem precisas, exa- tas e reprodutíveis.")
67
Os experimentos de recuperação devem ser
realizados comparando-se os resuldatos de amos- tras extraidas a três concentrações diferentes ( alta, média e baixa ) com padrões não extraidos que representam 100%.
 com padrões não extraidos que. representam 100%.")
68
Para o estudo da recuperação:
Emprega-se três níveis de concentração: baixo, que pode ser o LOQ; médio e alto, que estejam compreendidos dentro do intervalo de limite especificado do procedimento analítico. 5 á 10 determinações em replicata para cada nível de concentrações nominais pré-estabelecidas das amostras de controle de qualidade. * Mehta, 1989; Causon, R e FDA – Guideline for Industry / Bio- analytical Method Validation final draft ,1998 e 2001

69
Estabilidade A Resolução RDC Nº 84 de 19 de março de
2002 e FDA, 1997 , estabelecem que que o estudo da estabilidade deve ser considerada durante a fase de desenvolmento analítico. Neste sentido, os estudos de estabilidade foram classificados em:

70
Curta duração: Congelamento e descongelamento: - 70 ou - 20°C por 3 ciclos; recongelamento de hs; Condições de Análise: °C, tempo máximo de análise do lote; Condições de Auto Sampler: 25°C; tempo máximo de análise do lote;

71
Média duração: temperatura - 20 °C; Avaliar no tempo zero, um, dois quatro, oito e dezesseis dias de armazenamento. Longa duração; deve compreender no tempo entre a data da primeira coleta das amostras e a data da análise da última amostra 03 temperaturas de estocagem,4, - 20 e - 70ºC

72
Soluções-padrão; Deve ser avaliada à temperatura ambiente por no mínimo 6 horas. Se a solução estoque e padrão interno são refrigeradas ou congeladas por um período de quatorze dias,

73
Considerações Preparar três á cinco determinações das concentrações de controle de qualidade (baixa, média, e alta) juntamente com o padrão interno nas amostras biológicas e comparar nas condições determinadas com as mesmas amostras recém preparadas. Um composto é considerado estável se sua decomposição não ultrapassar 20% da sua concentração inicial ou não ultrapassar o limite estabelecido no protocolo de validação . O prazo de validade é estabelecido com base neste parâmetro
 juntamente com o padrão interno nas amostras biológicas e comparar nas condições determinadas com as mesmas amostras recém preparadas. Um composto é considerado estável se sua decomposição não ultrapassar 20% da sua concentração inicial ou não ultrapassar o limite estabelecido no protocolo de validação . O prazo de validade é estabelecido com base neste parâmetro.")
74
Critérios de Aceitabilidade
Precisão: Os CV% do inter-day para os QCs é 15%, e 20% para o LOQ QC, avaliados em um mínimo de 3 lotes. Exatidão: A média dos valores do inter-day devem ser menores que 15% do valor nominal dos QCs, e não desviar mais que 20% para LOQ QC. Sensibilidade: O menor ponto da curva de calibração aceitável como limite de quantificação (LOQ) do método é aquele com CV% 20% (inter-day).
 do método é aquele com CV% 20% (inter-day).")
75
Especificidade: Respostas de picos interferentes no mesmo tempo de retenção do analíto devem ser menores que 20% da resposta de um LOQ. Respostas de picos interferentes no mesmo tempo de retenção do padrão interno (I.S.) devem ser menores que 5% da resposta de um I.S. utilizado nos estudos.
 devem ser menores que 5% da resposta de um I.S. utilizado nos estudos.")
76
Estabilidade: Teste de Longo-Prazo, Curto-Prazo, Freezer and Thaw, Soluções de Estoque e Auto-Sampler são propostos no SOP e variam de estudo para estudo

77
Guia da Validação Obtenção do Método Analítico
Obtenção do Material Biológico Obtenção do Plasma Referência Bibliográfica Não disponível Disponível Congelamento Desenvolvimento do Método Adequação Recebimento Pré - Validação Armazenamento Método Validado Estabilidade

78
Pré - Validação Condição Analítica Definida Interferência
Sem Interferência Seletividade < 60% > 60% Recuperação > 15% < 20% Linearidade Limite de Quantificação > 20% < 15% > 15% Precisão < 15% Parâmetros de Aceitabilidade O.K Estabilidade > 15% < 15% Exatidão Restrições

79
Redefinir tempo de Estudo de Estocagem
Estabilidade Não compatível tempo Curta duração Compatível tempo Não compatível tempo Redefinir tempo de Estudo de Estocagem Média duração Compatível tempo Longa duração Iniciar o Estudo Não compatível tempo

80
Aplicação do Método Validado Obtenção do Material Biológico
O.K Inicio do Estudo Aplicação do Método Validado Voluntários Obtenção do Material Biológico Corrida do Lote Obtenção do Plasma Curvas Amostras C.Qs Congelamento O.K O.K O.K Recebimento APROVADO Armazenamento Estabilidade RELATÓRIO

Apresentações semelhantes


















































>")







