Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Electromiografia II Aula VIII: Polineuropatias Docente: Rui Braga

2
Polineuropatias Hereditárias

3
Polineuropatias Polineuropatia ou neuropatia periférica são termos que descrevem o síndrome clínico que se caracteriza por fraqueza, perda sensitiva e deterioração dos reflexos, causado por lesões difusas dos nervos periféricos. Podem ocorrer em qualquer idade Os sintomas podem-se instalar progressivamente ao longo de anos, meses e em alguns casos em horas.

4
Polineuropatias A maioria das polineuropatia afectam ambas as funções motoras e sensitivas. Os sintomas das PNP incluem acroparestesias, dores, fraqueza e perda de sensibilidade. A dor pode ser espontânea ou induzida por estimulação da pele. Na maioria dos casos a fraqueza acomete mais a musculatura distal, que podem levar a paralisia dos músculos intrínsecos das mãos e dos pés com pulso ou pé pendente.

5
Polineuropatias Os reflexos tendinosos poderão estar ausentes.
Nas PNP severas os pacientes podem ficar quadriplégicos e dependentes de um ventilador. A perda de sensibilidade cutânea poderá apresentar uma distribuição em meia ou em luva.

6
Polineuropatias Poderá haver afecção de todos os tipos de sensibilidades, ou poderá haver uma deterioração selectiva da função das grandes fibras (sensibilidade proprioceptiva e vibratória) ou das pequenas fibras (sensibilidade térmica e dolorosa). Poderá haver envolvimento do sistema nervoso autónomo causando miose, hipotensão ortostática, sintomas esfincterianos, impotência e anomalias vasomotoras.
 ou das pequenas fibras (sensibilidade térmica e dolorosa). Poderá haver envolvimento do sistema nervoso autónomo causando miose, hipotensão ortostática, sintomas esfincterianos, impotência e anomalias vasomotoras.")
7
Caso clínico Um homem de 23 anos de idade apresenta uma história com alguns anos de evolução de fraqueza progressiva dos tornozelos e movimentos desajeitados. O exame físico revelou que o paciente tinha fraqueza distal nos membros superiores e inferiores, com o pé pendente, dedos de martelo e pés bastante arqueados. Ligeira perda da sensibilidade vibratória e táctil e arreflexia. Apresentava nervos volumosos e palpáveis.

8
Caso clínico O estudo EMG revelou:
Amplitudes dos CMAP e SNAP reduzidas. Prolongamento das latências distais e das respostas-F e velocidades de condução substancialmente reduzidas (bem abaixo de 50% do limite inferior da normalidade). Registaram-se poucas diferenças na morfologia do PAMC por estimulação proximal e distal (amplitude, duração e morfologia). Não se registou evidência de dispersão temporal dos potenciais ou bloqueio de condução parcial. Na EMG de agulha obtiveram-se alterações neurogénicas crónicas na musculatura distal.
. Registaram-se poucas diferenças na morfologia do PAMC por estimulação proximal e distal (amplitude, duração e morfologia). Não se registou evidência de dispersão temporal dos potenciais ou bloqueio de condução parcial. Na EMG de agulha obtiveram-se alterações neurogénicas crónicas na musculatura distal.")
9
Caso clínico

10
Caso clínico Estes achados providenciam evidencias electrodiagnósticas de uma neuropatia sensorio-motora moderadamente severa, do tipo desmielinizante, com uma ligeira sobreposição de degeneração axonal. A marcada diminuição das velocidades de condução e ausência de dispersão temporal ou bloqueio de condução parcial é mais consistente com uma neuropatia hereditária.

11
Caso clínico O exemplo clássico de uma neuropatia caracterizada por uma mielinopatia periférica uniforme é a neuropatia sensorio-motora hereditária tipo I (Doença Charcot-Marrie-Tooth tipo I). CMT I é uma neuropatia hipertrófica autossómica dominante, que se apresenta por um início insidioso de fraqueza distal e perda de sensibilidade. O diagnóstico é clinicamente sugestivo pelos achados de nervos volumosos, fraqueza distal com dedos de pés em forma de martelo e pé cavo, alterações da sensibilidade vibratória e táctil e hiporreflexia.
. CMT I é uma neuropatia hipertrófica autossómica dominante, que se apresenta por um início insidioso de fraqueza distal e perda de sensibilidade. O diagnóstico é clinicamente sugestivo pelos achados de nervos volumosos, fraqueza distal com dedos de pés em forma de martelo e pé cavo, alterações da sensibilidade vibratória e táctil e hiporreflexia.")
12
Caso clínico Apresenta achados electrodiagnósticos característicos tais como: Velocidades de condução marcadamente reduzidas em todos os nervos testados, podendo ser da ordem dos 25 m/s ou menos. As VC encontram-se tipicamente 70% abaixo do limite inferior da normalidade. Habitualmente não se observa grande dispersão temporal dos potenciais, nem bloqueios de condução parciais devido ao envolvimento uniforme de todas as fibras. A maioria dos pacientes apresenta também pelo menos algum grau de perda axonal que por vezes poderá ser severa. A EMG de agulha demonstra recrutamento reduzido dos PUM, fibrilações e ondas positivas e PUM de grande amplitude e duração, polimórficos, reflectindo reínervação crónica.

13
Polineuropatias hereditárias

14
Doença de Charcot-Marie-Tooth
A doença de Charcot-Marie-Tooth é a doença neuromuscular hereditária mais comum que afecta os nervos periféricos. É uma doença neurológica hereditária, caracterizada por uma degeneração lenta e progressiva dos músculos dos pés, pernas, mãos e antebraços, e uma diminuição da sensibilidade nos membros, dedos das mãos e dos pés. Os seus sintomas podem variar muito, desde sintomatologia frustre até deformidades severas.

15
Doença de Charcot-Marie-Tooth
Um dos primeiros sinais de CMT é geralmente a presença de um pé bastante arqueado e alterações da marcha. Outros sintomas podem incluir anomalias ósseas nos pés tais como grandes arcadas e dedos em forma de martelo, problemas funcionais das mãos e do equilíbrio, perda de reflexos osteotendinosos, por vezes diminuição da capacidade auditiva ou mesmo surdez. Podem surgir em alguns pacientes escolioses.

16
Doença de Charcot-Marie-Tooth
Esta doença habitualmente não é fatal, podendo mesmo não prejudicar a qualidade de vida do paciente, não afecta as capacidades intelectuais, embora, nos casos mais extremos, dificuldades respiratórias relacionadas com CMT poderão levar à morte. Esta doença é hereditária com heterogeneidade genética, na qual mutações de diferentes genes poderão produzir sintomas clínicos semelhantes. Existem diferentes tipos de CMT

17
Polineuropatias hereditárias
Charcot-Marrie-Tooth tipo I (a e b) (HSMN I) – Caracteristicamente desmielinizante, neuropatia autossomica dominante hipertrofica. Charcot-Marrie-Tooth tipo II (HSMN II) – Forma axonal da Atrofia muscular peroneal. Charcot-Marrie-Tooth tipo III (HSMN III) – Neuropatia hipertrofica da infância (Doença de Dejerine-Sottas) Charcot-Marrie-Tooth tipo IV (HSMN IV) – Neuropatia hipertrofica (Refsum) associada ao excesso de acido Fitanico.
 (HSMN I) – Caracteristicamente desmielinizante, neuropatia autossomica dominante hipertrofica. Charcot-Marrie-Tooth tipo II (HSMN II) – Forma axonal da Atrofia muscular peroneal. Charcot-Marrie-Tooth tipo III (HSMN III) – Neuropatia hipertrofica da infância (Doença de Dejerine-Sottas) Charcot-Marrie-Tooth tipo IV (HSMN IV) – Neuropatia hipertrofica (Refsum) associada ao excesso de acido Fitanico.")
18
Charcot-Marrie-Tooth
Tradicionalmente, a patofisiologia da doença de CMT foi categorizada em dois processos: um predominantemente desmielinizante com velocidades de condução reduzidas (CMTI) e outro predominantemente axonal resultando em potenciais de baixa amplitude (CMTII)
 e outro predominantemente axonal resultando em potenciais de baixa amplitude (CMTII)")
19
Charcot-Marrie-Tooth tipo I
Na CMT tipo I o início dos sintomas dá-se habitualmente na primeira década, menos frequentemente na segunda década e raramente depois daí, ocasionalmente desde nascença. Dá-se uma fraqueza distal progressiva, máxima nos membros inferiores, acompanhada por arreflexia. A atrofia muscular inicialmente envolve a musculatura do nervo peroneal e posteriormente a coxa e os membros superiores.

20
Charcot-Marrie-Tooth tipo I
Poderá estar presente um tremor postural nos membros superiores Ocorre perda sensitiva distal: afectando todas as variedades e é de severidade variável. Deformidades nos pés, usualmente pé cavo, dedos em forma de garra e posição de equinovarus são frequentes.

21
Charcot-Marrie-Tooth tipo I

22
Charcot-Marrie-Tooth tipo I

23
Charcot-Marrie-Tooth tipo I
Podem-se desenvolver escolioses em pacientes com desenvolvimento precoce da sintomatologia. Alguns pacientes desenvolvem paralisia diafragmática, com falência respiratória ou cardíaca. A clássica configuração de cegonha da perna desenvolve-se apenas raramente na forma hipertrófica da doença. O pé pendente bilateral causa uma característica dificuldade da marcha.

24
Charcot-Marrie-Tooth tipo I
O paciente apresenta parestesias, disestesias e dores associadas às deformidades dos pés Tipicamente a sintomatologia inclui: nervos palpáveis, perda da sensibilidade vibratória e postural, diminuição da sensibilidade cutânea e diminuição dos reflexos, inicialmente nos tornozelos e mais tarde difusamente. A doença progride lentamente ao longo de várias décadas.

25
Charcot-Marrie-Tooth tipo I
A atrofia muscular e a fraqueza podem incapacitar o doente, mas nem sempre. Estudos histológicos revelam aumento do calibre dos nervos periféricos, com desmielinização segmentar e remielinização e atrofia axonal.

26
Charcot-Marrie-Tooth tipo I
A maioria das famílias demonstram uma inerência da doença autossómica dominante, embora também se observem casos autossómicos recessivos. Pacientes com esta última forma tendem a ser mais severamente afectados. É sabido que a maioria das famílias com HSMN I autossomica dominante, apresentam uma mutação no cromossoma 17 – Esta forma foi designada por HSMN Ia.

27
Charcot-Marrie-Tooth tipo I
Noutras familias, mas muito menos frequentemente, um gene anormal localiza-se no braço proximal do cromossoma 1 – Esta forma foi designada por HSMN Ib

28
EMG Charcot-Marrie-Tooth tipo I
As velocidades de condução apresentam-se acentuadamente baixas difusamente e de uma forma uniforme. As formas recessivas raras têm umas velocidades mais baixas do que as dominantes. As velocidades de condução motora em membros de famílias afectadas são cerca de metade dos indivíduos normais, variando entre 15 e 35 m/s com uma média de 25 m/s, nos nervos mediano e cubital.

29
EMG Charcot-Marrie-Tooth tipo I
Os potenciais de acção nervosos sensitivos compostos estão habitualmente ausentes ou dispersos e de baixa amplitude, e com velocidades de condução severamente reduzidas. As latências motoras distais podem-se encontrar aumentadas A explicação major para a redução da velocidade de condução nervosa é a desmielinização e remielinização, mas a perda das fibras largas e rápidas e a possível atrofia axonal também podem contribuir.

30
EMG Charcot-Marrie-Tooth tipo I
A lentificação da condução na HSMN I é uniforme e não se observam bloqueios de condução ou dispersão temporal significativa. Este factor é importante para distinguir pacientes com polineuropatia desmielinizante inflamatória crónica, nos quais, as velocidades de condução tendem a não ser concordantes nos diferentes nervos ou nos diferentes segmentos nervosos, nos quais se observam grandes dispersões temporais.

31
Charcot-Marrie-Tooth tipo II
A clínica da CMT tipo I difere da CMT tipo II na idade de iniciação da sintomatologia que é máxima na segunda década de vida e menos frequentemente na primeira década. Contudo, muitos pacientes, desenvolvem sintomas inicialmente na primeira década de vida, mas na maioria dos casos atrasa-se até à idade adulta, por vezes, até mesmo até à sétima década de vida. Comparativamente com a CMT I, há um menor envolvimento dos membros superiores e os músculos das pernas são mais severamente afectados., simultaneamente com os músculos do compartimento antero-lateral da perna.

32
Charcot-Marrie-Tooth tipo II
Apresentam fraqueza selectiva da musculatura da perna com envolvimento limitado dos membros superiores. Dá-se uma perda quase total do volume muscular abaixo do joelho dando às pernas uma aparência de pernas de cegonha. Tremor e ataxia e arreflexia difusa são habitualmente menos evidentes, assim como a severidade da perda de sensibilidade. As deformidades nos pés também são menos acentuadas, devendo-se este factor provavelmente ao desenvolvimento da doença mais tardio.

33
Charcot-Marrie-Tooth tipo II
Os pacientes não apresentam hipertrofia dos nervos. Alguns indivíduos podem apresentar algum tremor das mãos, mas muito menos comummente do que nos pacientes com CMT I.

34
EMG Charcot-Marrie-Tooth tipo II
EMG de agulha demonstra um recrutamento reduzido com evidência de reinervação. As velocidades de condução motoras são menos severamente diminuídas comparativamente com CMT I, podendo estas encontrar-se próximo do limite da normalidade ou ligeiramente diminuídas. A amplitude dos potenciais de acção sensitivos compostos é reduzida, particularmente nos membros inferiores, onde muitas das vezes os potenciais se encontram ausentes. PAMC também reduzidos.

35
Polineuropatia hipertrófica de Dejerine-Sottas (HSMN III)
Trata-se de uma forma de neuropatia sensorio-motora desmielinizante hereditária autossomica recessiva muito severa. Os nervos afectados encontram-se engrossados, com desmielinizações segmentares e com a mielina a envolver o nervo extremamente delgada. Os sintomas surgem na infância, com atraso no desenvolvimento das capacidades motoras, especialmente na marcha.

36
Polineuropatia hipertrófica de Dejerine-Sottas (HSMN III)
Os achados clínicos incluem: pés cavos, cãimbras, incoordenação, escolioses, fraqueza, perda de sensibilidade e paralisias dos nervos abducente e facial. Na maioria os adultos apresentam paraparésia e ataxia severa, requerendo o uso de cadeira de rodas. Pacientes com esta patologia tem uma maior incidência de ataxia, arreflexia e hipertrofia dos nervos do que os pacientes com CMT I. Apresenta umas velocidades de condução motoras e sensitivas caracteristicamente baixas, por vezes inferiores a 5 m/s.

37
Neuropatia Ataxica Hereditária de Refsum (HSMN tipo IV)
Trata-se de uma patologia rara que é transmitida através de um gene que pode ser dominante ou recessivo Caracteristicamente apresentam alterações no tracto olivo-cerebelar, células dos cornos anteriores da medula e nervos periféricos. A clínica típica apresenta surdez, anosmia, cegueira nocturna, retinite pigmentosa, sinais cerebelosos e nistagmo.

38
Neuropatia Ataxica Hereditária de Refsum (HSMN tipo IV)
O envolvimento dos nervos periféricos provoca dores e fraqueza muscular nas pernas, hiporreflexia e hipotonia e diminuição da sensibilidade vibratória e cinestesica. Um defeito metabólico na oxidação de ácidos gordos de cadeia longa leva a uma aumento sérico de ácido fitánico que, por razões desconhecidas leva a uma neuropatia hipertrofica

39
Polineuropatia Amiloidotica Familiar (Paramiloidose)
Em 1939 o Dr. Corino de Andrade, Director do Serviço de Neurologia do Hospital de santo António, observa um doente com alterações sensitivas, motoras e disfunções autonômicas cuja classificação não se enquadrava em qualquer situação patológica até então conhecida e que lhe conta que na sua família e na sua terra, Póvoa de Varzim, existem muitos casos semelhantes. Doze anos depois, em 1952, Corino de Andrade descreve pela 1ª vez na Brain, a Polineuropatia Amiloidótica Familiar de tipo Português, também denominada de Paramiloidose ou vulgarmente chamada de “ doença dos Pezinhos”.

40
Polineuropatia Amiloidotica Familiar (Paramiloidose)
Actualmente a doença encontra-se incluída no grande grupo das amiloidoses hereditárias. Estas por sua vez englobam as polineuropatias amiloidóticas familiares que se classificam: • PAF tipo I, de Andrade ou tipo Português; • PAF tipo II, de Rukovina ou tipo Indiana; • PAF tipo III, de Van Allen ou tipo Iowa; • PAF tipo IV, de Meretoja ou tipo Filandês

41
Polineuropatia Amiloidotica Familiar (Paramiloidose)
A Polineuropatia Amiloidótica Familiar (PAF) é uma doença neurológica, hereditária de transmissão autossómica dominante. È uma doença congénita que afecta o sistema nervoso periférico nas suas vertentes motora, sensitiva e autonômica. Inicia-se insidiosamente entre os 20 e 40 anos e conduz a um desfecho fatal após 10 a 15 anos de sofrimento. A sua etiopatogenia está relacionada com a produção pelo fígado, retina e plexos coroideus de uma proteína plasmática anómala, a TTR Met 30.
 é uma doença neurológica, hereditária de transmissão autossómica dominante. È uma doença congénita que afecta o sistema nervoso periférico nas suas vertentes motora, sensitiva e autonômica. Inicia-se insidiosamente entre os 20 e 40 anos e conduz a um desfecho fatal após 10 a 15 anos de sofrimento. A sua etiopatogenia está relacionada com a produção pelo fígado, retina e plexos coroideus de uma proteína plasmática anómala, a TTR Met 30.")
42
Polineuropatia Amiloidotica Familiar (Paramiloidose)
Todas as manifestações da PAF traduzem a degenerescência progressiva dos nervos Todas as fibras nervosas são atingidas e condicionam o quadro clínico da PAF: - Fibras Sensitivas (Perda progressiva da sensibilidade, primeiro a dor e temperatura, depois táctil, vibratória e articular); - Fibras Motoras (Atrofia muscular, Abolição dos reflexos tendinosos, Marcha em steppage) - Fibras Autonômicas (Perturbações oculares, perturbações cardiovasculares, perturbações gastrointestinais, perturbações genito-urinárias), etc.
; - Fibras Motoras (Atrofia muscular, Abolição dos reflexos tendinosos, Marcha em steppage) - Fibras Autonômicas (Perturbações oculares, perturbações cardiovasculares, perturbações gastrointestinais, perturbações genito-urinárias), etc.")
43
Polineuropatia Amiloidotica Familiar (Paramiloidose)
No estadio final, o doente encontra-se acamado ou numa cadeira de rodas, apresentando de uma forma geral, fraqueza muscular, atrofias, e caminhando inexoravelmente para a morte por caquexia e/ou infecções intercorrentes. A Paramiloidose é uma doença incurável e progressiva em todas as dimensões – biológica, psicológica, sociológica, cultural e espiritual

44
Polineuropatia Amiloidotica Familiar (Paramiloidose)
Um gene, localizado no cromossoma 18, sofreu um dia uma mutação, passando a transmitir uma informação errada. Vários órgãos ( fígado, plexos coroideus, retina) fabricam em consequência uma proteína diferente da normal. Esta proteína anómala é uma variante da transtirretina ( TTR) A TTR met 30, deposita-se sob a forma de uma substância anormal, a substância amilóide, em quase todos os tecidos do organismo. É uma doença congénita que afecta o sistema nervoso periférico nas suas vertentes motora, sensitivas e autonómica. O portador do gene pode desenvolver a paramiloidose.
 fabricam em consequência uma proteína diferente da normal. Esta proteína anómala é uma variante da transtirretina ( TTR) A TTR met 30, deposita-se sob a forma de uma substância anormal, a substância amilóide, em quase todos os tecidos do organismo. É uma doença congénita que afecta o sistema nervoso periférico nas suas vertentes motora, sensitivas e autonómica. O portador do gene pode desenvolver a paramiloidose.")
45
Polineuropatia Amiloidotica Familiar (Paramiloidose)
Medidas Terapêuticas: As alterações do ritmo cardíaco e da condução originam disritmias graves conduzindo inexoravelmente para a necessidade de pacemaker definitivo. Após o desenvolvimento laboratorial da Imunodepuração da TTR circulante no final dos anos 80, deu-se em início de 1993, a um programa de ensaios clínicos que consiste na filtração do sangue do doente, retirando a TTR met 30 através do uso de anticorpos monoclonais que se ligam à proteína anómala.

46
Polineuropatia Amiloidotica Familiar (Paramiloidose)
O transplante hepático è uma hipótese Terapêutica, iniciada por Holmgren na Suécia em 1991 e entre nós pela Unidade de Transplante Hepático do Curry Cabral em 1992 , com resultados encorajadores, sobretudo quando efectuado em fase relativamente precoce da doença. O objectivo do transplante hepático reside na não evolução da sintomatologia da paramiloidose, já que permite substituir o principal órgão produtor da proteína anómala. Não se trata de uma terapêutica curativa. Em finais de Dezembro de 2002, o CEAP tem uma população de 102 doentes transplantados nas várias unidades a nível nacional.

47
Polineuropatias associadas a condições médicas gerais

48
Polineuropatia diabética

49
Polineuropatia Diabética
A diabetes mellitus é uma das causas mais frequentes de neuropatia no mundo. Diz-se que um paciente tem polineuropatia diabética, quando apresenta sintomas e/ou sinais de disfunção dos nervos periféricos, quando este é diabético, após a exclusão de outras causas.

50
Causas da Polineuropatia Diabética
Embora se julgue que a hiperglicémia seja a base para o desenvolvimento da disfunção neurológica na diabetes, o mecanismo específico inerente à sua patogénese aínda não está claramente esclarecido. A manutenção dos níveis sanguíneos de glicose dentro dos limites normais, melhora a velocidade de condução nervosa motora, desacelera a progressão da neuropatia e, por vezes, reverte os sinais neuropaticos. Doença vascular periférica é comum nos diabéticos, mas não há uma correlação directa entre a severidade da patologia vascular e a polineuropatia.

51
Causas da Polineuropatia Diabética
Outras teorias defendem que a hiperglicémia causa um aumento dos niveis de glicose intracelular dos nervos, levando à saturação da via glicolitica normal. A glicose excedente vai intervir na via do poliol e é convertida em sorbitol e fructose por enzimas. A acumulação do sorbitol e fructose leva a uma redução do mioinositol do nervo, decremento da actividade da ATPase-Na+/K+, prejudicando o transporte axonal e a uma queda da funcionalidade do nervo, causando lentificação da velocidade de condução.

52
Polineuropatia diabética
Na diabetes Mellitus tipo I, a polineuropatia periférica ocorre tipicamente após muitos anos de hiperglicémia crónica prolongada. Contrariamente, na DM tipo II, a PNP poderá apresentar-se após apenas poucos anos de um pobre controlo de glicemia. Ocasionalmente, na DM II, a neuropatia diabética é descoberta em simultâneo na altura do diagnóstico.

53
Sintomas Polineuropatia diabética
A PNP diabética poderá apresentar-se por uma grande variedade de sintomas, sensitivos, motores e autonómicos. Os sintomas sensitivos poderão ser negativos ou positivos, difuso ou focal: Sintomas sensitivos negativos incluem entorpecimento, sensação de pés mortos, perda de equilibrio, especialmente com os olhos fechados, e analgesia que habitualmente conduz a lesões. Os sintomas positivos incluem dores com sensação de queimadura, sensação de choques eléctricos e hipersensibilidade ao toque.

54
Sintomas Polineuropatia diabética
Os sintomas motores podem causar fraqueza proximal, distal ou focal. Os sintomas motores distais incluem afecção da coordenação fina das mãos, incapacidade para abrir frascos ou girar chaves, marcha com batida dos pés, etc Sintomas de fraqueza proximal incluem dificuldade em descer e subir escadas, dificuldades em levantar-se a partir de uma posição sentada, quedas devidas a fraquejar dos joelhos e dificuldades em levantar os braços acima dos ombros.

56
Sintomas Polineuropatia diabética
Sintomas autonómicas poderão ser sudomotores (pele seca, falta de sudação, excesso de sudação), pupilares (pobre adaptação à escuridão, sensibilidade a luzes brilhantes), cardiovasculares, urinários (urgência urinária ou incontinência), gastrointestinal e sexual (disfunção eréctil e frigidez). Uma classificação geralmente aceite das neuropatias diabéticas dividem-nas de uma forma geral em neuropatias simétricas e assimétricas.
, pupilares (pobre adaptação à escuridão, sensibilidade a luzes brilhantes), cardiovasculares, urinários (urgência urinária ou incontinência), gastrointestinal e sexual (disfunção eréctil e frigidez). Uma classificação geralmente aceite das neuropatias diabéticas dividem-nas de uma forma geral em neuropatias simétricas e assimétricas.")
57
Neuropatia distal simétrica
É a neuropatia diabética mais comum. Sintomas simétricos crónicos envolvem as extremidades distais. Os nervos periféricos são afectados de uma forma comprimento-dependente, em que os nervos mais longos são afectados primariamente. As funções sensitivas, motoras e autonomicas são afectadas de diversos graus, coma disfunção sensitiva a predominar.

58
Neuropatia distal simétrica
Frequentemente apresentam parestesias e entorpecimento nocturnas que se iniciam habitualmente pelos dedos dos pés e ascendem em forma de meia ao longo do tempo. Quando os sintomas sensitivos atingem os joelhos, as mãos desenvolvem sintomas similares, progredindo proximalmente em forma de luva. Mais tarde desenvolve-se fraqueza dos músculos dos pés e diminuição dos reflexos dos tornozelos e dos joelhos.

59
Neuropatia distal simétrica
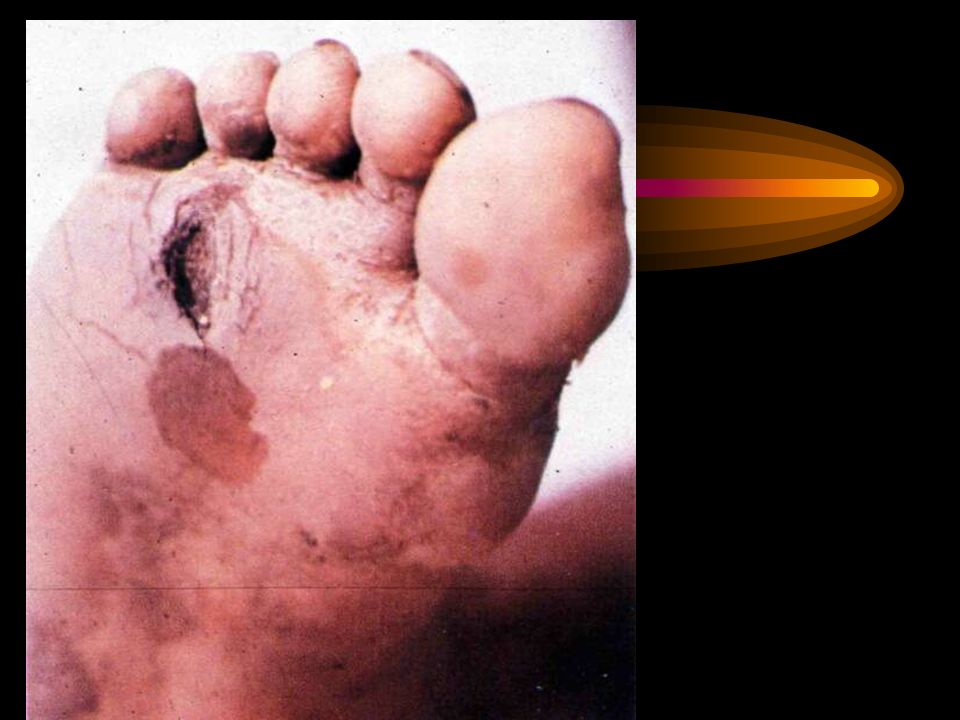
Perda da sensibilidade térmica e dolorosa por afecção das pequenas fibras, levando a úlceras nos pés. Alterações da sensibilidade proprioceptiva, vibratória e alterações da marcha por envolvimento das fibras largas. Alterações esfincterianas, atonia da bexiga e pupilas arreactivas poderão dever-se a disfunção aotonómica.

60
EMG na PNP diabética Deverão ser encontradas alterações ao nível das velocidades de condução sensitivo-motoras, mesmo que o paciente não apresenta clínica evidente de polineuropatia distal simétrica. As alterações iniciais deverão ser encontradas nos nervos sensitivos (sural, peroneal superficial, mediano). As velocidades de condução motoras não devem ser inferiores a 50% do limite normal. Se for deverá ser investigada outra causa de PNP.
. As velocidades de condução motoras não devem ser inferiores a 50% do limite normal. Se for deverá ser investigada outra causa de PNP.")
61
EMG na PNP diabética Os potenciais de acção sensitivo composto deverá apresentar amplitude reduzida, com potenciais mal formados ou mesmo ausentes, com lentificação da velocidade de condução. Não deverão estar presentes sinais de bloqueio de condução. Registo de PAMC de baixa amplitude, habitualmente nos peroniais.

62
EMG na PNP diabética Respostas F e reflexos H com latências prolongadas ou ausentes. Na EMG agulha regista-se nos músculos distais sinais de desnervação sobre a forma de fibrilações e ondas positivas. Potenciais de reinervação (grande amplitude e duração e polifasicos)
")
63
Polineuropatia alcoólica
Primariamente afecta aqueles que bebem uma grande quantidade de álcool, por um longo período de anos Habitualmente melhora com a abstinência Juntamente com o possível efeito tóxico que o álcool tem, deficiências nutricionais e mal-absorção também são factores importante

64
Polineuropatia alcoólica
Muitos pacientes alcoólicos apresentam uma deficiência em vitamina B1 e Tiamina, que por si só podem levar a achados clínicos semelhantes. Os sintomas clínicos habitualmente aparecem de uma forma insidiosa ao longo de semanas ou meses, mas por vezes de uma forma mais aguda, ao longo de alguns dias As primeiras queixas sensitivas habitualmente são dores distaIs, parestesias e disestesias, inicialmente nas pernas e posteriormente nos braços

65
Polineuropatia alcoólica
Sensação de queimadura nas extremidades Em casos mais avançados apresentam pés-pendentes bilateralmente, associados a atrofias musculares distais, envolvendo mais os flexores do que os extensores

66
Polineuropatia Urémica
Existe uma variedade de polineuropatias que resultam do efeito complexo da falência renal ao nível dos neurónios periféricos Esta PNP desenvolve-se em pacientes com insuficiência renal crónica severa ou em pacientes em hemodialise crónica

67
Polineuropatia Urémica
O uso de drogas neurotoxicas, tais como a nitrofurantoina podem contribuir para a destruição nervosa. Os sintomas clínicos desenvolvem-se habitualmente de uma forma abrupta. Os membros inferiores tendem a ser afectados mais cedo e de forma mais acentuada que os membros superiores Alguns pacientes apresentam sindrome das pernas irrequietas como sintomas Com a hemodialise habitualmente os pacientes melhoram.

Apresentações semelhantes









, e que pode levar a incapacitação funcional.>")









