Carregar apresentação
A apresentação está carregando. Por favor, espere

2
Síndrome de Down aspectos neurológicos
José Salomão Schwartzman Prof. Titular Pós-graduação em Distúrbios do desenvolvimento da Universidade Presbiteriana Mackenzie

4
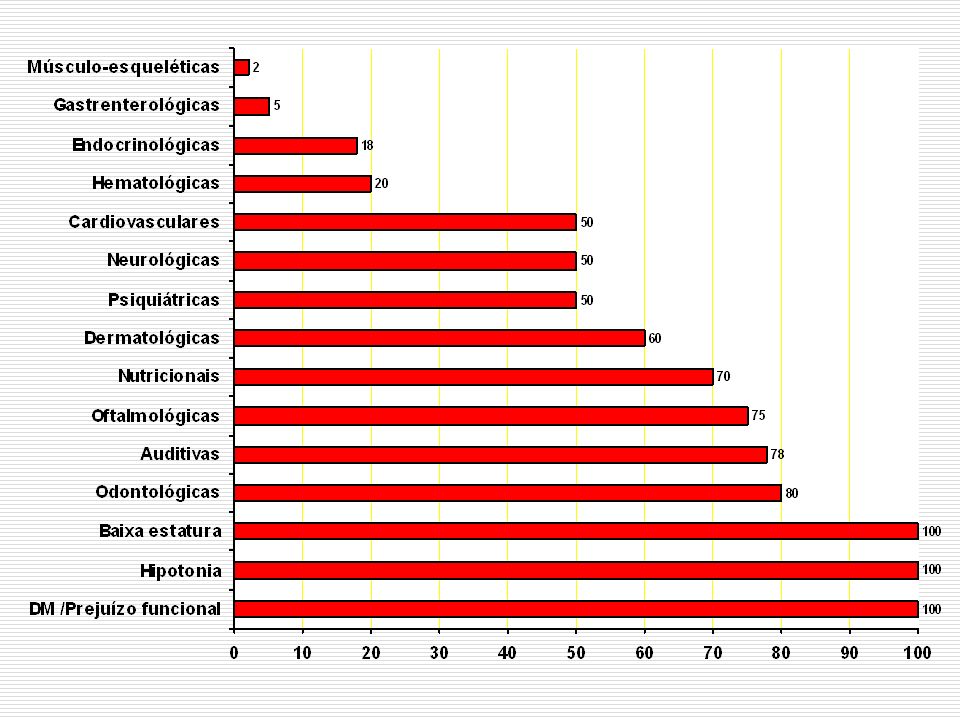
Alterações neurológicas
o sistema nervoso central (SNC) apresenta anormalidades estruturais e funcionais que determinarão as disfunções neurológicas sempre presentes estas disfunções variam bastante quanto às suas manifestações e grau de severidade
 apresenta anormalidades estruturais e funcionais que determinarão as disfunções neurológicas sempre presentes. estas disfunções variam bastante quanto às suas manifestações e grau de severidade.")
5
Alterações neurológicas
os achados neuropatológicos sugerem que na SD existam defeitos neurais e sinápticos pré-natais patologias encefálicas mais evidentes não estão presentes na vida intra-uterina mas surgem precocemente após o nascimento

6
Alterações neurológicas
desenvolvimento pré-natal do SNC os achados são, em geral, pouco significativos e discordantes Sylvester (1983) observou, em um feto de 18 semanas de gestação, que o encéfalo era menor e o hipocampo menor e malformado Schmidt-Sidor et al. (1990) compararam os achados em 17 fetos com SD com 17 fetos não Down, não tendo observado diferenças significativas no que se refere ao crescimento e maturação cerebral; o cerebelo e tronco cerebral eram do mesmo tamanho; o desenvolvimento dos córtices cerebrais e cerebelares era similar
 observou, em um feto de 18 semanas de gestação, que o encéfalo era menor e o hipocampo menor e malformado. Schmidt-Sidor et al. (1990) compararam os achados em 17 fetos com SD com 17 fetos não Down, não tendo observado diferenças significativas no que se refere ao crescimento e maturação cerebral; o cerebelo e tronco cerebral eram do mesmo tamanho; o desenvolvimento dos córtices cerebrais e cerebelares era similar.")
7
Alterações neurológicas
vários estudos demonstram que o cérebro de crianças com SD recém-nascidas ou logo após este período é quase que indistinguível do de crianças normais valores normais no que se refere à forma do encéfalo e crânio, peso encefálico, tamanho proporcional dos lobos, tamanho do cerebelo e tronco cerebral bem como a emergência da maior parte dos sistemas de neurotransmissores

8
síndrome de Down controle
encéfalos de recém-natos com SD (a,b) não diferem, praticamente, de encéfalos de recém-natos não Down (mod. de Wisniewski,1990 síndrome de Down controle
 não diferem, praticamente, de encéfalos de recém-natos não Down. (mod. de Wisniewski,1990. síndrome de Down. controle.")
9
síndrome de Down controle
após os 3-5 meses de idade, as diferenças anatômicas entre crianças com (a,c) e sem SD (b,d) são evidentes: diminuição dos lobos frontais, achatamento dos pólos occipitais e menor tamanho do tronco cerebral e cerebelo (mod. de Wisniewski,1990) síndrome de Down controle
 e sem SD (b,d) são evidentes: diminuição. dos lobos frontais, achatamento dos pólos occipitais e menor. tamanho do tronco cerebral e cerebelo (mod. de Wisniewski,1990) síndrome de Down. controle.")
10
Alterações neurológicas
Wisniewski (1990) acompanhou 780 crianças com SD, do nascimento até os 5 anos de idade ao nascimento, a forma do encéfalo era similar ao de crianças não Down enquanto que o peso situava-se nas faixas inferiores da normalidade entre 3 e 6 meses de idade ocorria uma desaceleração do crescimento do encéfalo que podia ser avaliado pelo perímetro cefálico em 69% dos casos, entre os 7 e 12 meses de idade, o peso do encéfalo mostrava-se abaixo dos valores normais
 acompanhou 780 crianças com SD, do nascimento até os 5 anos de idade. ao nascimento, a forma do encéfalo era similar ao de crianças não Down enquanto que o peso situava-se nas faixas inferiores da normalidade. entre 3 e 6 meses de idade ocorria uma desaceleração do crescimento do encéfalo que podia ser avaliado pelo perímetro cefálico. em 69% dos casos, entre os 7 e 12 meses de idade, o peso do encéfalo mostrava-se abaixo dos valores normais.")
11
Alterações neurológicas
a referida desaceleração mostrava-se mais acentuada nas meninas e fortemente associada com a presença de doenças cardíacas congênitas e malformações gastrointestinais

12
Alterações neurológicas
por volta do sexto mês de vida diversas diferenças importantes começam a ficar aparentes porem o perfil das mesmas não é uniforme encontramos um retardo na mielinização em cerca de 25% dos casos que é global inicialmente tornando-se mais evidente nos tratos que conectam os lobos temporais e frontais (necropsias entre 2 meses e 6 anos) nesses casos, a mielinização mostrava-se normal ao nascimento
 nesses casos, a mielinização mostrava-se normal ao nascimento.")
13
Alterações neurológicas
estudos morfométricos demonstram que a densidade neuronal na área frontal, temporal e occipital é menor ao nascimento em indivíduos com SD (Wisniewski et al., 1984, 1986, 1993; Wisniewski, 1990) já foram descritas diminuição na densidade sináptica, no comprimento pré-sináptico e na área média por contato sináptico bem como anomalias na morfologia das sinapses (Petit et al., 1984; Wisniewski et al., 1986)
 já foram descritas diminuição na densidade sináptica, no comprimento pré-sináptico e na área média por contato sináptico bem como anomalias na morfologia das sinapses (Petit et al., 1984; Wisniewski et al., 1986)")
14
Densidade neuronal no córtex visual
idade em anos neurônios/mm3 SD controles Densidade neuronal no córtex visual densidade neuronal média na SD é menor do que nos controles desde o nascimento

15
Alterações neurológicas
encontramos uma redução de 10%-50% no peso do encéfalo (Wisniewski et al., 1986) o peso do encéfalo de adultos normais varia de 1200g-1500g enquanto que de indivíduos com SD varia de 700g-1100g o perímetro cefálico de crianças com SD mostra-se 2 a 3 desvios padrão abaixo dos valores normais em crianças com menos de 5 anos de idade
 o peso do encéfalo de adultos normais varia de 1200g-1500g enquanto que de indivíduos com SD varia de 700g-1100g. o perímetro cefálico de crianças com SD mostra-se 2 a 3 desvios padrão abaixo dos valores normais em crianças com menos de 5 anos de idade.")
16
redução significativa na substância branca:
Estudo morfométrico de adultos com síndrome de Down não demenciados (White, Alkire e Haier, 2003) redução significativa na substância cinzenta: cerebelo giro do cíngulo lobo frontal esquerdo medial giro temporal direito superior/médio hipocampo (região CA2/CA3) esquerdo redução significativa na substância branca: tronco cerebral inferior
 redução significativa na substância cinzenta: cerebelo. giro do cíngulo. lobo frontal esquerdo medial. giro temporal direito superior/médio. hipocampo (região CA2/CA3) esquerdo. redução significativa na substância branca: tronco cerebral inferior.")
17
Funções do córtex pré-frontal
é particularmente importante nas funções executivas tem importância na memória explícita (episódica)
")
18
áreas límbicas: emoções e memória
giro do cíngulo núcleos mediais núcleo anterior núcleo accumbens área septal núcleo basal de Meynert corpo mamilar hipocampo córtex órbito-frontal corpo amigdalóide giro para-hipocampal áreas límbicas: emoções e memória

20
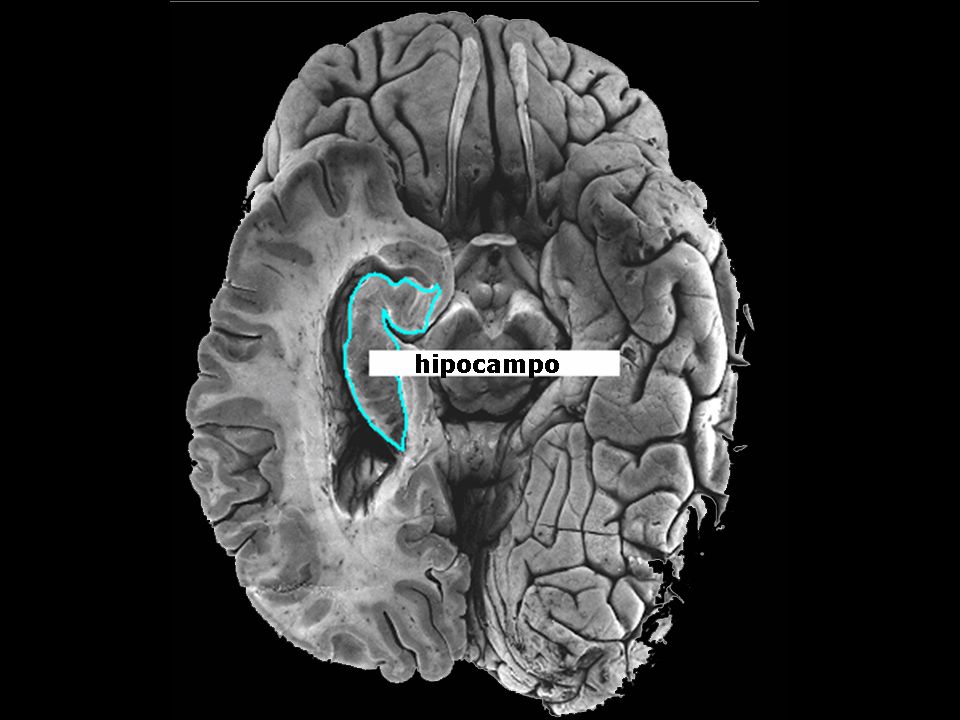
controle síndrome de Down
secções coronais do hipocampo de um feto não Down (a) e de um feto com SD (b): o hipocampo no feto Down é menor e menos pregueado havendo, ainda, várias outras diferenças histológicas entre as duas amostras (Sylvester, 1983) controle síndrome de Down
 e. de um feto com SD (b): o hipocampo no feto Down é menor e. menos pregueado havendo, ainda, várias outras diferenças. histológicas entre as duas amostras (Sylvester, 1983) controle. síndrome de Down.")
21
Funções hipocampo o hipocampo está envolvido na cognição espacial, na aprendizagem em geral e na consolidação de informações já apreendidas não está envolvido na aprendizagem de categorias e conceitos nem na aprendizagem de habilidades

22
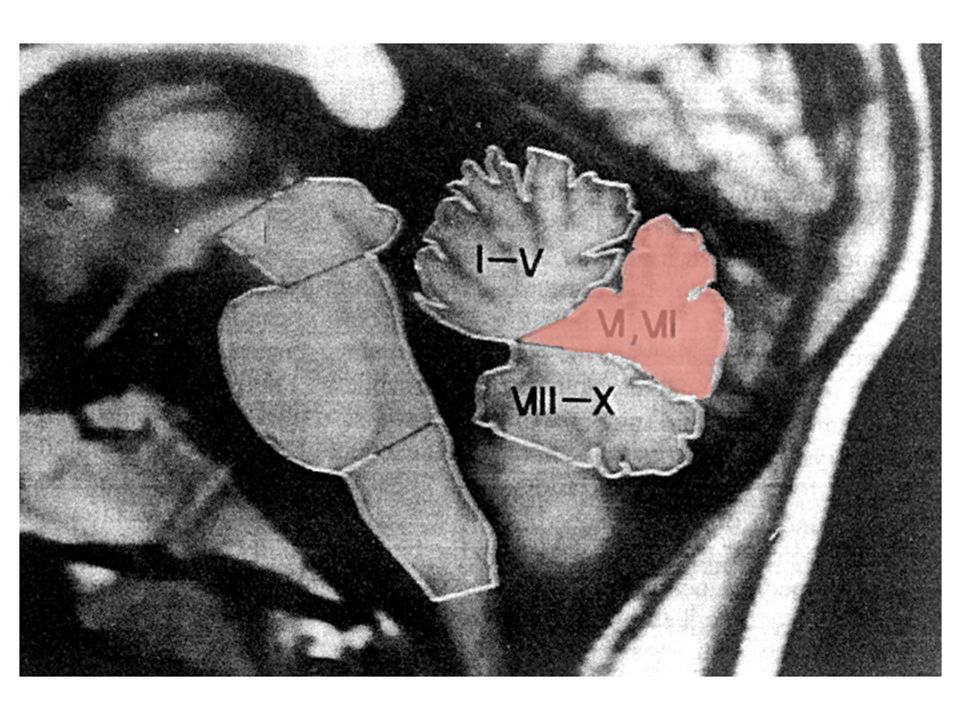
cerebelo

23
Funções do cerebelo não são totalmente conhecidas
está envolvido em tarefas motoras parece ser importante para a aquisição de respostas condicionadas

24
redução significativa na substância branca:
Estudo morfométrico de adultos com síndrome de Down não demenciados (White, Alkire e Haier, 2003) redução significativa na substância cinzenta: cerebelo giro do cíngulo lobo frontal esquerdo medial giro temporal direito superior/médio hipocampo (região CA2/CA3) esquerdo redução significativa na substância branca: tronco cerebral inferior
 redução significativa na substância cinzenta: cerebelo. giro do cíngulo. lobo frontal esquerdo medial. giro temporal direito superior/médio. hipocampo (região CA2/CA3) esquerdo. redução significativa na substância branca: tronco cerebral inferior.")
25
Anormalidades neuroanatômicas seletivas na SD e seus correlatos cognitivos (Raz et al., 1995)
os autores estudaram (ressonância nuclear magnética da cabeça) 13 adultos SD e 12 controles grupo SD apresentou, em comparação com o grupo controle: redução dos hemisférios cerebrais e cerebelares redução dos lóbulos VI, VII e VIII do vermis cerebelar redução da região ventral da ponte e dos corpos mamilares redução da formação hipocampal diminuição do córtex pré-frontal dorsolateral, região anterior do giro cíngulo, córtices temporal inferior e parietal, substância branca parietal e córtex pericalcarino aumento do giro para-hipocampal
 13 adultos SD e 12 controles. grupo SD apresentou, em comparação com o grupo controle: redução dos hemisférios cerebrais e cerebelares. redução dos lóbulos VI, VII e VIII do vermis cerebelar. redução da região ventral da ponte e dos corpos mamilares. redução da formação hipocampal. diminuição do córtex pré-frontal dorsolateral, região anterior do giro cíngulo, córtices temporal inferior e parietal, substância branca parietal e córtex pericalcarino. aumento do giro para-hipocampal.")
26
6 7 8

27
Anormalidades neuroanatômicas seletivas na SD e seus correlatos cognitivos (Raz et al., 1995)
hipoplasia do vermis cerebelar pode ser observado no autismo e síndrome do X-frágil achados similares já foram descritos em outras anormalidades cromossômicas

29
Anormalidades neuroanatômicas seletivas na SD e seus correlatos cognitivos (Raz et al., 1995)
o aumento do giro para-hipocampal decorre, provavelmente, de displasia cortical localizada, uma desordem da migração neuronal que ocorre nos primeiros estágios da gestação observaram relação inversa entre o tamanho do giro para-hipocampal e os níveis cognitivos gerais

30
áreas límbicas: emoções e memória
giro do cíngulo núcleos mediais núcleo anterior núcleo accumbens área septal núcleo basal de Meynert corpo mamilar hipocampo córtex órbito-frontal corpo amigdalóide giro para-hipocampal áreas límbicas: emoções e memória

31
fazem parte do fenótipo da SD:
Síndrome de Down: uma perspectiva biocomportamental de uma desordem genética (Nadel, 2003) os sistemas neurais mais comprometidos parecem ser a formação hipocampal, o córtex pré-frontal e o cerebelo fazem parte do fenótipo da SD: dificuldades na aquisição da informação (aprendizagem) dificuldades na manutenção a longo prazo e recuperação da informação (memória)
 os sistemas neurais mais comprometidos parecem ser a formação hipocampal, o córtex pré-frontal e o cerebelo. fazem parte do fenótipo da SD: dificuldades na aquisição da informação (aprendizagem) dificuldades na manutenção a longo prazo e recuperação da informação (memória)")
32
Outras alterações neurológicas
algum grau de disfunção neuromotora está presente na SD: hipotonia, hiporreflexia miotática e diminuição dos reflexos primitivos disgenesia do córtex cerebral e cerebelo bem como retardo na mielinização durante os primeiros anos de vida, constituem o substrato neurológico primário da referida disfunção atrasos nos marcos do desenvolvimento motor são perceptíveis já durante os primeiros meses de vida

33
Outras alterações neurológicas
há considerável variação individual no grau e padrão de disfunção motora entre vários fatores que podem interferir nesta variabilidade está a presença de alguma condição médica tal como doença cardíaca congênita, crises convulsivas, hipotireoidismo, grau de hipotonia etc.

34
perímetro cefálico (NCHS) em meninas, meninos e
indivíduos com SD (mod. Rogers & Coleman, 1992)
")
35
evolução do perímetro cefálico de um rapaz de 14 anos com SD

36
evolução do perímetro cefálico de uma menina de 14 anos com SD

37
Alterações hipotalâmicas
algumas crianças com SD apresentam diminuição do hormônio de crescimento ((HC) possivelmente secundário a uma disfunção hipotalâmica uma boa resposta à administração do HC com aumento do crescimento somático e do perímetro cefálico já foi observada (Annerén et al., 1986; Wisniewski et al., 1989; Torrado e t al., 1991; Castells et al., 1992, 1993)
 possivelmente secundário a uma disfunção hipotalâmica. uma boa resposta à administração do HC com aumento do crescimento somático e do perímetro cefálico já foi observada (Annerén et al., 1986; Wisniewski et al., 1989; Torrado e t al., 1991; Castells et al., 1992, 1993)")
38
Capacidades intelectuais
desde as descrições originais tem se considerado a deficiência mental como uma das características mais constantes da SD na maioria dos trabalhos publicados, as crianças obtém, em testes formais de inteligência, pontuações no QI que variam entre 20 e 85

39
Capacidades intelectuais
a SD é a causa de 17% dos casos de deficiência mental nos USA Moore (1973) avaliando 2750 indivíduos com SD encontrou 2 com QI acima de 85 e 7 com QI entre 70 e 84 a disfunção cognitiva observada nestes pacientes pode ser heterogênea há evidências de declínio no QI que pode ser observado à partir dos 6 meses de idade
 avaliando 2750 indivíduos com SD encontrou 2 com QI acima de 85 e 7 com QI entre 70 e 84. a disfunção cognitiva observada nestes pacientes pode ser heterogênea. há evidências de declínio no QI que pode ser observado à partir dos 6 meses de idade.")
40
Capacidades intelectuais
pacientes com SD demonstram prejuízos significativos em, praticamente, todas as áreas do funcionamento intelectual especialmente na compreensão verbal e raciocínio(Rondal et al., 1987) em comparação com controles com DM pontuam menos em testes de orientação, coordenação óculo-manual, memória visual, nomeação de objetos e conhecimentos gerais (Thase et al., 1984)
 em comparação com controles com DM pontuam menos em testes de orientação, coordenação óculo-manual, memória visual, nomeação de objetos e conhecimentos gerais (Thase et al., 1984)")
41
Capacidades intelectuais desenvolvimento mental na síndrome de Down com mosaico
Fishler e Koch (1991) avaliaram o desenvolvimento intelectual de 30 pacientes com SD com mosaico e 30 com trissomia 21 mosaico: 16 do sexo masculino e 14 do feminino idades variando entre 2 e mais de 18 anos pareados por idade cronológica e sexo com o grupo trissomia testes utilizados na avaliação: escalas de desenvolvimento de Gesell escala de Kuhlmann-Binet escala de inteligência de Stanford-Binet escala de internacional de performance de Leiter teste de vocabulário Peabody teste de inteligência para adultos Weschler WAIS-R teste de inteligência para crianças Weschler WISC-R
 avaliaram o desenvolvimento intelectual de 30 pacientes com SD com mosaico e 30 com trissomia 21. mosaico: 16 do sexo masculino e 14 do feminino. idades variando entre 2 e mais de 18 anos. pareados por idade cronológica e sexo com o grupo trissomia. testes utilizados na avaliação: escalas de desenvolvimento de Gesell. escala de Kuhlmann-Binet. escala de inteligência de Stanford-Binet. escala de internacional de performance de Leiter. teste de vocabulário Peabody. teste de inteligência para adultos Weschler WAIS-R. teste de inteligência para crianças Weschler WISC-R.")
42
Capacidades intelectuais desenvolvimento mental na síndrome de Down com mosaico
resultados grupo trissomia: média do QI foi de 52 (18 – 78) dp 14.6 grupo mosaico: média do QI foi de 64 (43 – 92) dp 13.8 média o QI no sexo masculino: 64 média do QI no sexo feminino: 67 não houve correlação entre o QI e a porcentagem de células normais
 dp grupo mosaico: média do QI foi de 64 (43 – 92) dp média o QI no sexo masculino: 64. média do QI no sexo feminino: 67. não houve correlação entre o QI e a porcentagem de células normais.")
43
grupo mosaico grupo trissomia idade em anos QI/QD

44
Capacidades intelectuais
avaliações do QI em indivíduos com SD têm mostrado aumentos significativos nas última décadas os dados referentes à avaliação da inteligência em pacientes com SD devem ser interpretados cuidadosamente o declínio descrito anteriormente pode ser apenas aparente

45
Estabilidade das habilidades cognitivas (Carr, 2005)
estudos longitudinais de mostram um declínio do QI na população geral com a idade: o declínio é: precoce e rápido no caso da inteligência não verbal (habilidade fluida) tardio e mais lento na inteligência verbal (habilidade cristalizada)
 tardio e mais lento na inteligência verbal (habilidade cristalizada)")
46
Estabilidade das habilidades cognitivas (Carr, 2005)
estudos em indivíduos com SD diferem em dois aspectos: as habilidades cristalizadas (verbais) parecem manter-se menos bem do que em indivíduos da população geral o declínio nas habilidades fluidas é menos consistente do que o que ocorre na população geral
 parecem manter-se menos bem do que em indivíduos da população geral. o declínio nas habilidades fluidas é menos consistente do que o que ocorre na população geral.")
47
Estabilidade das habilidades cognitivas (Carr, 2005)
“inteligência cristalizada” em indivíduos com SD: grupos SD com idades entre 20 e 40 anos mostram um declínio nas provas verbais variando de 17% a 29% na população geral a queda observada entre os 20 e os 80 anos foi de 7% aparentemente as habilidades verbais parecem ser menos cristalizadas em pessoas com SD do que em indivíduos da população geral

48
Aprendizagem na SD não sabemos até que ponto a experiência pode modificar o cérebro normal não sabemos se as possibilidades de plasticidade cerebral que ocorrem em indivíduos normais ocorrem da mesma forma em indivíduos com SD não sabemos quais modificações cerebrais induzidas por várias formas de estimulação podem se traduzir em melhoras comportamentais ou cognitivas não sabemos quais modificações podem ser benéficas ou não

49
Aprendizagem na SD Ohr e Fagen (1991, 1993) estudaram a habilidade de crianças com SD para aprender comportamentos motores à partir da contingência entre seus próprios movimentos (chutar com os pés) e reforço crianças com 3 meses de idade (todo o grupo) mostraram-se inteiramente normais nesta tarefa, incluindo a aprendizagem inicial, rapidez de aquisição e retenção crianças com 9 meses (parte do grupo) mostraram-se prejudicadas nesta tarefa concluíram haver uma diminuição na condicionabilidade de crianças com SD após os 6 meses de idade
 estudaram a habilidade de crianças com SD para aprender comportamentos motores à partir da contingência entre seus próprios movimentos (chutar com os pés) e reforço. crianças com 3 meses de idade (todo o grupo) mostraram-se inteiramente normais nesta tarefa, incluindo a aprendizagem inicial, rapidez de aquisição e retenção. crianças com 9 meses (parte do grupo) mostraram-se prejudicadas nesta tarefa. concluíram haver uma diminuição na condicionabilidade de crianças com SD após os 6 meses de idade.")
50
Aprendizagem na SD apresentam diferenças e não simplesmente atrasos no desenvolvimento no período pré-verbal: comunicação gestual atenção comportamentos de aprendizagem o desenvolvimento das habilidades comunicativas não segue o mesmo curso do observado em crianças normais na idade em que se espera que iniciem atenção conjunta não pedem objetos ou ajuda com objetos sua atenção é mais focada em pessoas e menos em objetos demonstram menos interesse e prazer no aprender (Sigman, 1999)
")
51
Aprendizagem na SD estudos sobre a aprendizagem da linguagem demonstram que o prejuízo na aquisição da mesma pode ser bastante severa, principalmente nos domínios da sintaxe e da fonologia mas existem casos nos quais esta capacidade pode ser considerada normal (Tager-Flusberg, 1999; Thordardottir et al., 2002; Vicari et al., 2002) bebês com SD mostram habilidades pré-linguísticas normais (balbucio e imitação) porém podem demonstrar déficits no uso de solicitações não verbais (Mundy et al., 1988) as dificuldades desproporcionais no desenvolvimento da linguagem parecem ser independentes dos processos de aprendizagem e de memória parece haver um problema de instabilidade de aquisição
 bebês com SD mostram habilidades pré-linguísticas normais (balbucio e imitação) porém podem demonstrar déficits no uso de solicitações não verbais (Mundy et al., 1988) as dificuldades desproporcionais no desenvolvimento da linguagem parecem ser independentes dos processos de aprendizagem e de memória. parece haver um problema de instabilidade de aquisição.")
52
Síndromes genéticas com comportamentos
associados, possivelmente, específicos s. Prader-Willi hiperfagia s. Lesch-Nyhan auto-agressão extrema s. de Down habilidades visuais > habilidades auditivas receptivas s. Smith-Magenis tendência à introduzir objetos nos orifícios corporais; auto-abraço s. Williams altas habilidades de linguagem e pobre funcionamento viso-espacial s. de Rett estereotipias manuais s. de Angelman marcha atáxica e episódios de riso s. cri-du-chat choro tipo “grito/miado de gato” na infância

53
síndrome de Down idade mental idade quando do teste Faces (Benton)
Linguagem Espacial idade quando do teste idade mental síndrome de Down

54
Faces (Benton) Linguagem idade mental Espacial idade quando do teste
linguagem, processamento facial e espacial na síndrome de Williams

55
desenhos livres de casas: pacientes com SW e SD pareados por QI
grama meu quarto de dormir e das minhas irmãs lugar de ver TV piscina janelas porta casa de 2 andares calçada tethado Síndrome de Williams Síndrome de Down desenhos livres de casas: pacientes com SW e SD pareados por QI

56
Epilepsia prevalência de 1,4% (Caraballo et al., 2004)
Prasher (1996) em adultos com a SD encontrou 15,9% com epilepsia Johansen et al. (1996), referem freqüência de 17% em portadores da SD com idade entre 14 e 60 anos
 em adultos com a SD encontrou 15,9% com epilepsia. Johansen et al. (1996), referem freqüência de 17% em portadores da SD com idade entre 14 e 60 anos.")
57
Epilepsia manifestações epilépticas ocorrem em 6,4% dos indivíduos com SD (Stafstrom et al., 1991); este número é superior ao observado na população geral, mas é inferior ao encontrado em outras condições que cursam com deficiência mental (20% a 50%) quanto à idade de início, há 2 picos de incidência maior, sendo um na infância e outro na idade adulta mais avançada, quando se associam com o início da demência tipo-Alzheimer (Pueschel et al., 1991)
; este número é superior ao observado na população geral, mas é inferior ao encontrado em outras condições que cursam com deficiência mental (20% a 50%) quanto à idade de início, há 2 picos de incidência maior, sendo um na infância e outro na idade adulta mais avançada, quando se associam com o início da demência tipo-Alzheimer (Pueschel et al., 1991)")
58
Síndrome de Down e síndrome de West
Silva et al. (1996) descreveram esta associação em 14 casos acompanhados por 4 anos e meio idade de início do quadro: entre 4 e 18 meses 7 pacientes apresentaram outros tipos de manifestações epilépticas crises mioclônicas crises atônicas crises tônico-clônicas crises de ausência
 descreveram esta associação em 14 casos acompanhados por 4 anos e meio. idade de início do quadro: entre 4 e 18 meses. 7 pacientes apresentaram outros tipos de manifestações epilépticas. crises mioclônicas. crises atônicas. crises tônico-clônicas. crises de ausência.")
59
Síndrome de West caracterizada por 3 componentes: os espasmos
a alteração eletroencefalográfica denominada de hipsarritmia deterioração ou deficiência mental

60
Síndrome de West os espasmos iniciam-se, em geral, durante o primeiro ano de vida com o pico de incidência sendo entre os 4 e os 10 meses deficiência mental está presente, antes do início dos espasmos em cerca de 68% a 85% dos casos a SW é classificada em sintomática e criptogenética ou idiopática

63
estudados 20 pacientes com SW e SD 13 receberam tratamento habitual
Espasmos infantis na Síndrome de Down: uma boa resposta à vitamina B6 (Caraballo et al., 2004) estudados 20 pacientes com SW e SD 13 receberam tratamento habitual ACTH, ácido valpróico, vigabatrina 7 pacientes tratados com vitamina B6 4 meninas 3 meninos
 estudados 20 pacientes com SW e SD. 13 receberam tratamento habitual. ACTH, ácido valpróico, vigabatrina. 7 pacientes tratados com vitamina B6. 4 meninas. 3 meninos.")
64
7 pacientes tratados com vitamina B6
Espasmos infantis na Síndrome de Down: uma boa resposta à vitamina B6 (Caraballo et al., 2004) 7 pacientes tratados com vitamina B6 4 haviam sido tratados com vigabatrina e 3 com ACTH sem sucesso as drogas foram suspensas e a vitamina B6 por via oral foi iniciada 2 semanas após a suspensão da medicação antiepiléptica em 5 e 4 semanas após nas outras 2
 7 pacientes tratados com vitamina B6. 4 haviam sido tratados com vigabatrina e 3 com ACTH sem sucesso. as drogas foram suspensas e a vitamina B6 por via oral foi iniciada 2 semanas após a suspensão da medicação antiepiléptica em 5 e 4 semanas após nas outras 2.")
65
Espasmos infantis na Síndrome de Down: uma boa resposta à vitamina B6 (Caraballo et al., 2004)
alguns pacientes que não responderam bem ao primeiro curso de piridoxina responderam a um segundo curso a droga foi mantida por 24 meses a 30 meses e descontinuada em seguida, sem a ocorrência de recidivas nenhum dos pacientes apresentou efeitos colaterais durante o tratamento

66
a dose oral de B6 variou de 200 a 400 mg dia (25-50 mg/kg/dia)
Espasmos infantis na Síndrome de Down: uma boa resposta à vitamina B6 (Caraballo et al., 2004) a dose oral de B6 variou de 200 a 400 mg dia (25-50 mg/kg/dia) controle dos espasmos ocorreu em duas semanas em 4 dos pacientes os EEGs se normalizaram assim que os espasmos deixaram de ser observados
 a dose oral de B6 variou de 200 a 400 mg dia (25-50 mg/kg/dia) controle dos espasmos ocorreu em duas semanas em 4 dos pacientes. os EEGs se normalizaram assim que os espasmos deixaram de ser observados.")
67
Piridoxina (B6) efeitos colaterais descritos (Benninger et al., 1993):
inapetência períodos de irritabilidade e choro possível dor abdominal vômitos apatia gastrite hemorrágica

68
Epilepsia em indivíduos com SD, a síndrome dos espasmos infantis (síndrome de West) tem, em geral, evolução mais benigna do que em crianças não Down uma vez que há menor regressão psicomotora e menor tendência para a evolução para outros tipos de epilepsia um tipo de manifestação epiléptica similar à síndrome de West foi descrita em crianças com SD que receberam triptofano, amino-ácido que foi utilizado no passado, na tentativa de melhorar a hipotonia destes pacientes
 tem, em geral, evolução mais benigna do que em crianças não Down uma vez que há menor regressão psicomotora e menor tendência para a evolução para outros tipos de epilepsia. um tipo de manifestação epiléptica similar à síndrome de West foi descrita em crianças com SD que receberam triptofano, amino-ácido que foi utilizado no passado, na tentativa de melhorar a hipotonia destes pacientes.")
69
Doença de Alzheimer a doença de Alzheimer (DA) é a mais comum causa de demência associada à idade avançada há indícios de que adultos com SD são mais propensos a apresentar a DA do que adultos não-Down os achados neuropatológicos observados na DA e nos pacientes com SD e demência são bastante similares

70
Doença de Alzheimer pacientes com SD desenvolvem os sinais neuropatológicos característicos da DA muito mais cedo do que indivíduos com Alzheimer sem a trissomia 21 placas senis e enovelados neurofibrilares podem ser observados nos cérebros de indivíduos com SD que morrem após os 40 anos de idade

71
Doença de Alzheimer sabemos, hoje, que o peptídeo beta-amilóide é um dos constituintes principais das placas neuríticas e dos enovelados neurofibrilares este peptídeo é um derivado da proteína beta-amilóide precursora (APP) existem evidências de que o peptídeo beta-amilóide desempenhe papel importante na patogenia da doença de Alzheimer e da síndrome de Down o gene APP localiza-se no cromossomo 21 e mutações deste gene foram observadas em algumas famílias com pessoas com DA
 existem evidências de que o peptídeo beta-amilóide desempenhe papel importante na patogenia da doença de Alzheimer e da síndrome de Down. o gene APP localiza-se no cromossomo 21 e mutações deste gene foram observadas em algumas famílias com pessoas com DA.")
72
Doença de Alzheimer a presença de placas neuríticas na SD seria decorrente da cópia extra do cromossomo 21 com a conseqüente overdose do gene APP já foram detectadas concentrações elevadas da APP no soro e tecido cerebral de pacientes com SD

73
Doença de Alzheimer experimentos em animais demonstraram que deposições de peptídeo beta-amilóide são neurotóxicos (Kowall et al., 1992) foram produzidos ratos transgênicos que expressam elevados níveis da APP humana e nos quais podem ser demonstradas alterações neuropatológicas típicas da DA (Games et al., 1995; LaFerla et al., 1995) este é um excelente modelo animal da DA que poderá trazer resultados importantes para a compreensão da patogênese e possivelmente terapia da DA e da SD
 este é um excelente modelo animal da DA que poderá trazer resultados importantes para a compreensão da patogênese e possivelmente terapia da DA e da SD.")
74
Doença de Alzheimer alterações histológicas características da DA estão presentes na maioria dos pacientes com SD que morrem após os 35 anos de idade muito embora em boa parte destes casos não haja, em vida, nenhuma evidência de quadros demenciais (Ropper e Williams, 1980) estudos de Hof et al., (1995) demonstram, entretanto, que o padrão das alterações neuropatológicas encontradas na SD, embora semelhante, não é idêntico ao observado em indivíduos idosos não-Down nem em indivíduos não-Down com DA
 estudos de Hof et al., (1995) demonstram, entretanto, que o padrão das alterações neuropatológicas encontradas na SD, embora semelhante, não é idêntico ao observado em indivíduos idosos não-Down nem em indivíduos não-Down com DA.")
75
encéfalo de paciente com síndrome de Down e
demência (mod. de Rogers e Coleman, 1992)
")
76
aumento dos ventrículos e atrofia da região do hipocampo e
ressonância nuclear magnética: cortes coronais de indivíduos com doença de Alzheimer (E) e com SD com sinais clínicos de demência (F) mostrando aumento dos ventrículos e atrofia da região do hipocampo e giro para-hipocampal (setas) (mod. de Kesslak et al., 1994
 e com SD com sinais clínicos de demência (F) mostrando. aumento dos ventrículos e atrofia da região do hipocampo e. giro para-hipocampal (setas) (mod. de Kesslak et al.,")
77
Doença de Alzheimer a prevalência na população geral é de 2% a 3% nos indivíduos acima dos 65 anos e possivelmente o dobro em indivíduos com mais de 80 anos na população de 69 pacientes com SD estudada por Lai e Williams (1989), a prevalência foi de 8% na idade de 49 anos e de mais de 75% entre aqueles acima de 60 anos
, a prevalência foi de 8% na idade de 49 anos e de mais de 75% entre aqueles acima de 60 anos.")
78
Doença de Alzheimer há evidências de que, após os 50 anos de idade, indivíduos com SD perdem habilidades ou “envelhecem” de forma mais rápida do que indivíduos com outras condições tais como deficiência mental, paralisia cerebral e epilepsia Zigman et al., (1987) estudaram as habilidades adaptativas de 2144 indivíduos com SD, comparando-os com um grupo constituído por 4172 pacientes não-Down com prejuízos
 estudaram as habilidades adaptativas de 2144 indivíduos com SD, comparando-os com um grupo constituído por 4172 pacientes não-Down com prejuízos.")
79
Doença de Alzheimer & SD
dados obtidos pelo estudo colaborativo EURODERM (1991): há um risco 2,7 vezes maior de desenvolvimento da DA se o indivíduo tem um parente de primeiro grau com SD há um risco aumentado para a ocorrência de demência entre mães de indivíduos com SD o risco encontrado foi de 5 vezes maior entre mães com idade abaixo de 35 anos e não se mostrou aumentado em mães com idade superior a 35 anos quando do nascimento do bebê com trissomia 21 estes dados sugerem que haja uma susceptibilidade genética conjunta para a DA e SD
: há um risco 2,7 vezes maior de desenvolvimento da DA se o indivíduo tem um parente de primeiro grau com SD. há um risco aumentado para a ocorrência de demência entre mães de indivíduos com SD. o risco encontrado foi de 5 vezes maior entre mães com idade abaixo de 35 anos e não se mostrou aumentado em mães com idade superior a 35 anos quando do nascimento do bebê com trissomia 21. estes dados sugerem que haja uma susceptibilidade genética conjunta para a DA e SD.")
80
Quadros psiquiátricos
indivíduos com SD podem apresentar problemas psiquiátricos Menolascino (1965) encontrou, entre 86 crianças com SD, 11 (13%) com “condições psiquiátricas” Gath & Gumley (1986) encontraram, entre 193 crianças e adolescentes com SD, 73 (38%) com algum quadro psiquiátrico Lund (1988) encontrou, entre 44 adultos com SD, 11 (25%) com problemas psiquiátricos
 encontrou, entre 86 crianças com SD, 11 (13%) com condições psiquiátricas Gath & Gumley (1986) encontraram, entre 193 crianças e adolescentes com SD, 73 (38%) com algum quadro psiquiátrico. Lund (1988) encontrou, entre 44 adultos com SD, 11 (25%) com problemas psiquiátricos.")
81
Quadros psiquiátricos
Myers & Pueschel (1991) estudaram 497 indivíduos com SD: 261 tinham menos de 20 anos 164 tinham 20 anos ou mais idade média de 19,4 anos variando entre 1 e 72 288 do sexo masculino
 estudaram 497 indivíduos com SD: 261 tinham menos de 20 anos. 164 tinham 20 anos ou mais. idade média de 19,4 anos variando entre 1 e do sexo masculino.")
82
Quadros psiquiátricos
quadros psiquiátricos observados: desordem do déficit de atenção comportamentos oposicionais agressividade ansiedade quadros depressivos prováveis quadros demenciais anorexia nervosa quadros fóbicos desordem de Tourette

83
Autismo infantil & SD autismo infantil tem sido descrito em pacientes com SD: Myers & Pueschel (1991): em 1% dos indivíduos Gath & Gumley (1986): em 10% dos pacientes Lund (1988) em 11,3% dos indivíduos estima-se a ocorrência dos distúrbios abrangentes em cerca de 13% dos indivíduos com SD e cerca de 3% a do autismo
: em 10% dos pacientes. Lund (1988) em 11,3% dos indivíduos. estima-se a ocorrência dos distúrbios abrangentes em cerca de 13% dos indivíduos com SD e cerca de 3% a do autismo.")
84
180 indivíduos com síndrome de Down, idades entre 5 – 19 anos
instrumento questionário ASQ 15,6% TID (ASQ) 5,58% autismo: 8 masc. e 2 fem. 10,05% TID não autismo: 9 masc., 9 fem. de 21 TID-ASQ: 14 TID–clínico de 10 não TID–ASQ: 14 não TID e 2 TID-clínico
 5,58% autismo: 8 masc. e 2 fem. 10,05% TID não autismo: 9 masc., 9 fem. de 21 TID-ASQ: 14 TID–clínico. de 10 não TID–ASQ: 14 não TID e 2 TID-clínico.")
85
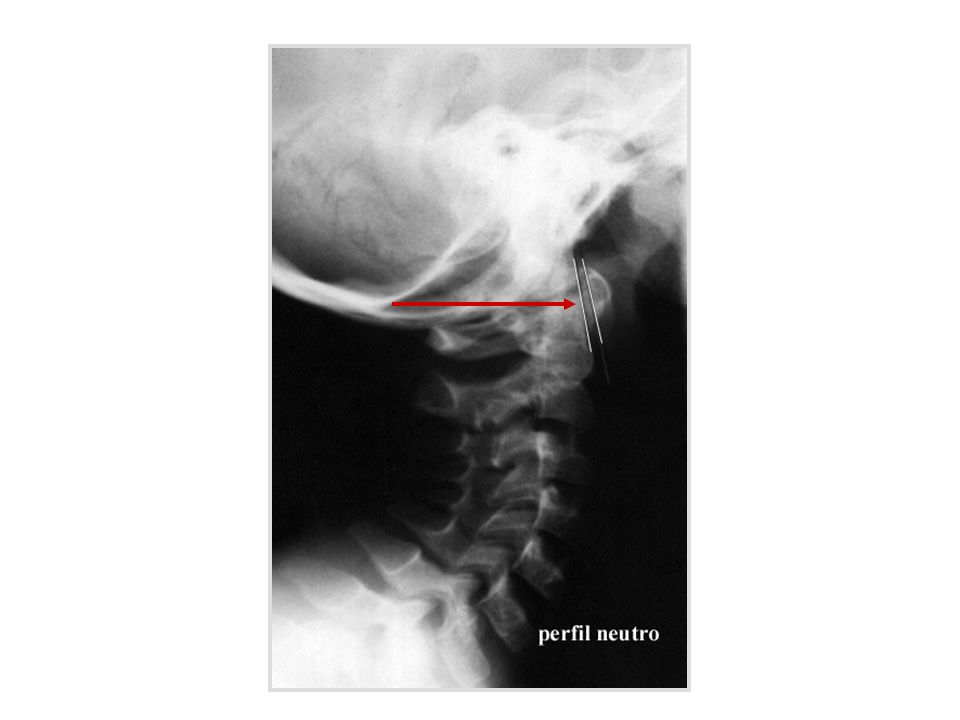
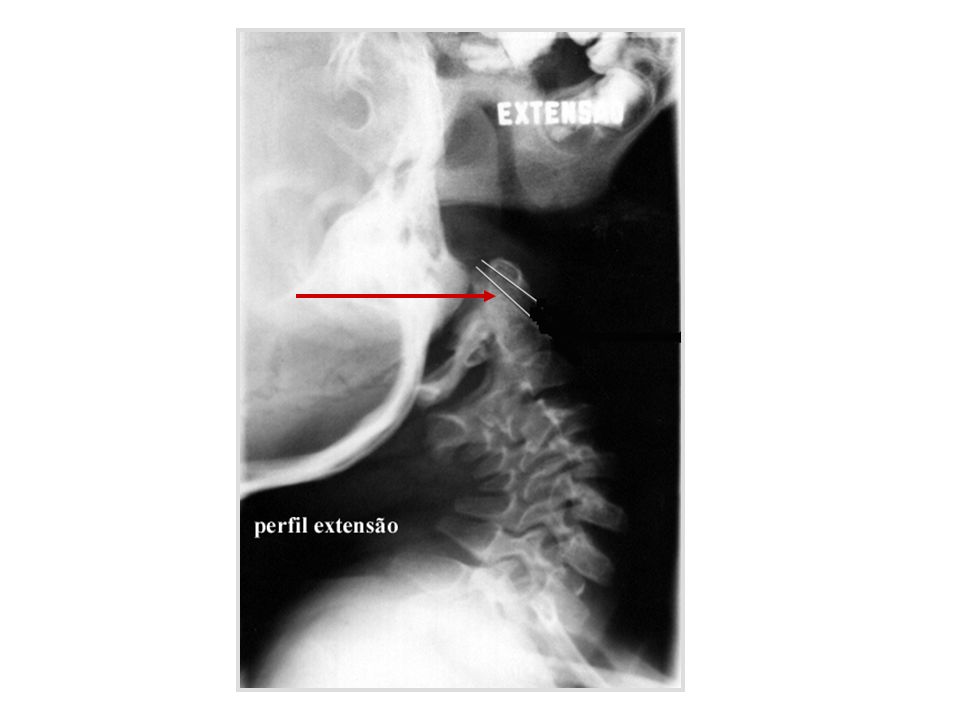
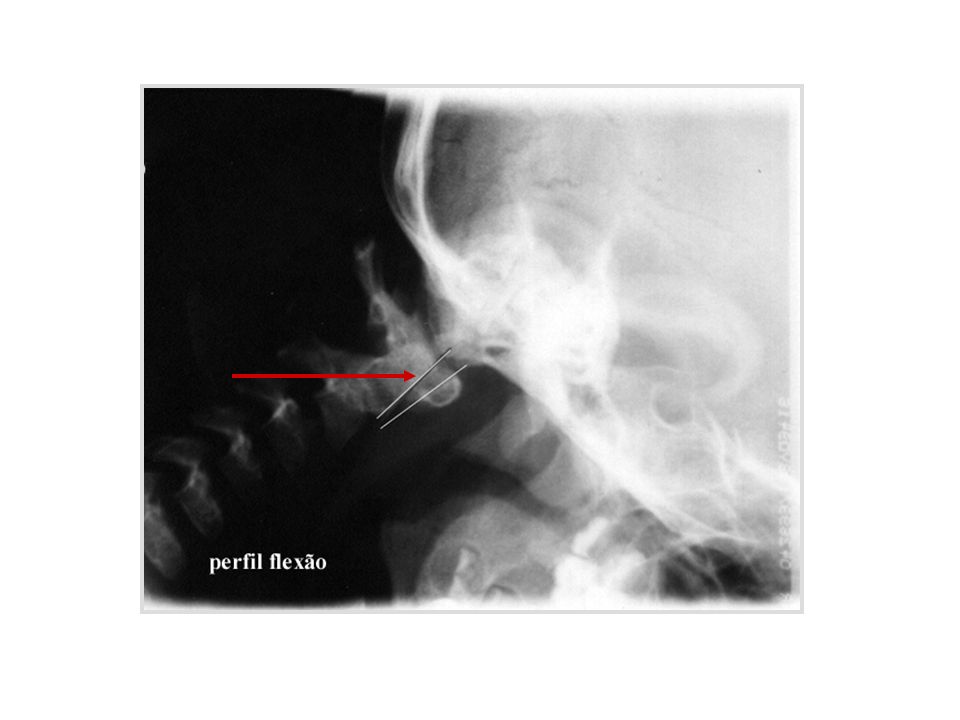
Instabilidade atlanto-axial
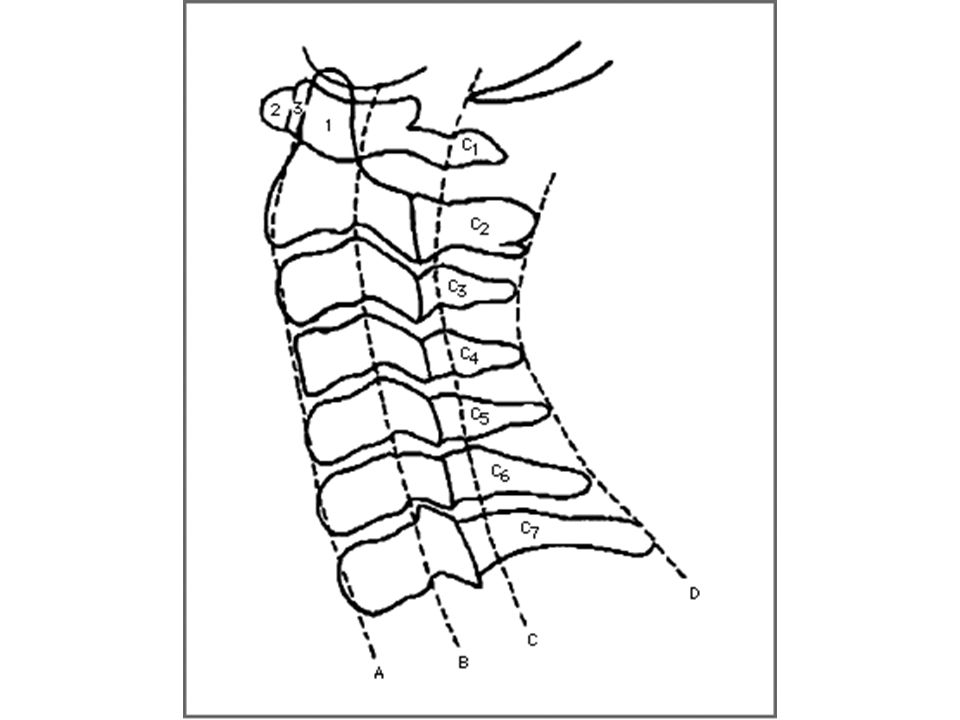
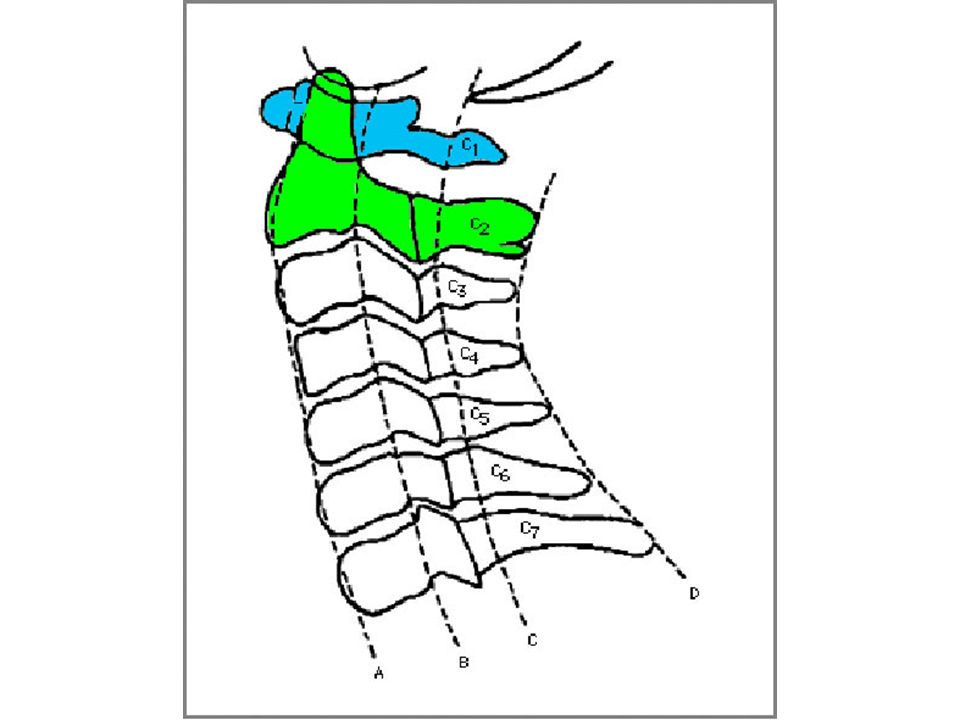
quando a distância A-A se modifica de forma significativa com a mudança de posição da coluna cervical sub-luxação atlanto-axial distância entre apófise anterior do odontóide e arco posterior do atlas maior de 5 mm causas: frouxidão ligamentar generalizada frouxidão do ligamento transverso alterações anatômicas da apófise odontóide hipoplasia displasia agenesia

88
Esquema da articulação atlanto-axial
odontóide arco anterior do atlas Esquema da articulação atlanto-axial

89
Instabilidade atlanto-axial
assintomática na maioria dos casos sintomática: sintomas agudos sintomas crônicos diagnóstico conduta

90
anatomia topográfica da região da articulação atlanto-axial

91
distância atlanto-axial
canal raquídeo C1 C2 C3 perfil neutro flexão extensão esquema da articulação atlanto-axial

95
Outros problemas....

96
hidrocefalia + anomalia de Dandy-Walker

97
hidrocefalia + anomalia de Dandy-Walker

98
hidrocefalia + anomalia de Dandy-Walker

99
siringomielia

100
hidrocefalia + anomalia de Dandy-Walker

101
Apnéia do sono obstrução das vias aéreas: estes fatores facilitam:
obstrução da faringe hipotonia da musculatura faríngea dimensões reduzidas da traquéia secreções estes fatores facilitam: apnéia do sono hipoxemia hipertensão pulmonar

102
Apnéia do sono apnéia do sono fatores predisponentes
hipotonia músculos da faringe hipotonia da língua redução das dimensões da cavidade oral redução das dimensões da faringe hipertrofia amígdalas e adenóides palato estreito hipoplasia da região média da face

103
Apnéia do sono incidência estimada: entre 30% e 60%
estudo de Shott et al.(2006) 65 crianças com idades entre 2 e 4 anos 56 completaram polissonografia noturna 57% evidências de síndrome da apnéia obstrutiva de sono 69% dos pais não relataram problemas de sono em seus filhos destes, 54% apresentavam polissonogramas anormais
 65 crianças com idades entre 2 e 4 anos. 56 completaram polissonografia noturna. 57% evidências de síndrome da apnéia obstrutiva de sono. 69% dos pais não relataram problemas de sono em seus filhos. destes, 54% apresentavam polissonogramas anormais.")
104
Apnéia do sono estudo de Shott et al.(2006): os autores concluem que:
devido à alta incidência da apnéia obstrutiva de sono em crianças jovens com síndrome de Down devido à pobre percepção dos pais a respeito de distúrbios do sono em seus filhos é recomendável a realização de polissonografia, de rotina, entre os 3 e os 4 anos em toda criança com síndrome de Down

Apresentações semelhantes




















>")

