Carregar apresentação
A apresentação está carregando. Por favor, espere

1
INTRODUÇÃO AO ESTUDO DA PATOLOGIA
VANESSA CORRALO

2
Tecido sanguíneo É um fluido indispensável à vida, que circula pelo coração e vasos sanguíneos levando oxigênio e nutrientes aos tecidos e resíduos catabólicos inúteis para os pulmões, o fígado e os rins, onde são excretados dos rins.

3
Hematopoese Ocorre na medula óssea, linfonodos e baço.
Até pouco antes do nascimento o fígado constitui-se principal órgão de hematopoese. Nascimento- medula óssea Até a Puberdade- medula óssea avermelhada e hematopoeticamente ativa. Aos 18 anos, apenas as vértebras,costelas, esterno,épífises do úmero e fêmur-medula vermelha. Restante- gordurosa e inativa Aumento na demanda- gordurosa –vermelha (7-8 vezes)
")
4
Medula óssea

5
Composição do sangue

7
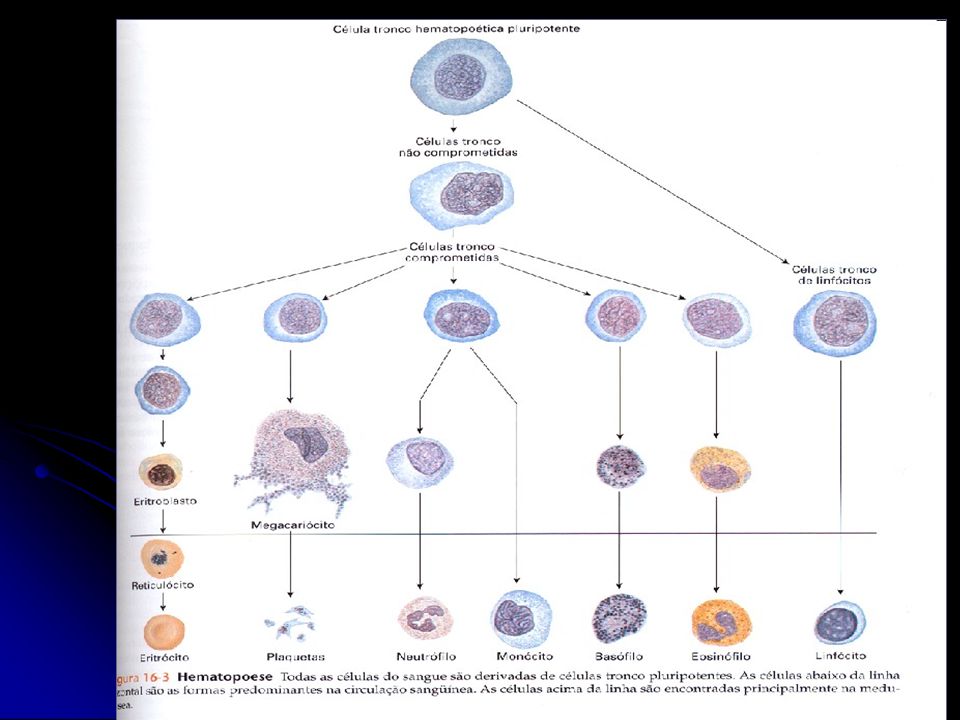
O hematócrito é a porcentagem (%) do volume total de sangue que é ocupada pelas células vermelhas.
Contagem de células vermelhas: em milhões de células por microlitro. Uma máquina conta as células de acordo com a quantidade de luz que passa por elas. A morfologia das células vermelhas: fornece diagnóstico de doenças.

8
Sangue Plasma Leucócitos Hemácias Esfregaço Hematócrito

9
Hemácias Funções Quantidade Tamanho

10
Leucócitos Tipos granulócitos neutrófilos agranulócitos linfócitos
eosinófilos basófilos agranulócitos linfócitos monócitos eosinófilo neutrófilo basófilo linfócito linfócito monócito

11
Plaquetas

12
Anemia

13
Definição Diz-se haver anemia quando a concentração da hemoglobina sanguínea diminui aquém de níveis arbitrados pela Organização Mundial de Saúde, são eles: 13 g/dL para homens 12 g/dL para mulheres 11 g/dL para gestantes e crianças entre 6 meses e 6 anos

14
Hemoglobina Localização Função Estrutura Tipos de Hb

15
Hemoglobina Localização: exclusiva dos eritrócitos
Função: transporte de O2 dos pulmões aos capilares dos tecidos .

16
Estrutura Hemeproteína
grupo protéico: globina + grupo prostético: heme cromoproteína Globina Tetrâmeros constituídos de 4 cadeias de polipepitídeo 2 e 2 (, ou ) Formam 2 dímeros: 11 e 22
 Formam 2 dímeros: 11 e 22.")
17
Tipos Normais de Hemoglobina
Hb A1: hemoglobina adulta; 2 cadeias alfa e 2 beta; = 95% do total de Hb Hb A2: 2 cadeias alfa e 2 delta; = 2% do total de Hb Hb F: principal Hb no feto e recém-nascido; 60% da Hb total até o 8º mês de gestação; substituída por Hb A1; menos de 5% no adulto; baixa afinidade por O2: liga-se fracamente ao 2,3-DPG Hb A1c : Hb A1 glicosilada; ligação da glicose a cadeia beta; 5 a 7% da Hb A1Diabete Mellitus

18
Anemia- Sinais e Sintomas
Palidez Fadiga Dor em membros inferiores Dispnéia aos esforços Taquicardia Angina Cefaléia Tonturas Irritabilidade

19
Classificação pela Patogênese
Perda sangüínea muito recente Anemia por carência de ferro (Anemia Ferropênica) Anemia por carência de Vitamina B12 e Ácido Fólico Anemia das doenças crônicas Anemias por defeitos genéticos Anemias por destruição periférica dos eritrócitos Anemias decorrentes de doenças da medula óssea
 Anemia por carência de Vitamina B12 e Ácido Fólico. Anemia das doenças crônicas. Anemias por defeitos genéticos. Anemias por destruição periférica dos eritrócitos. Anemias decorrentes de doenças da medula óssea.")
20
Perda Sanguínea Muito Recente Anemia Pós-Hemorrágica
Sangue remanescente é normal Rápida hemorragia: plasma reposto de 1 a 3 dias, porém há baixa concentração de hemácias

21
Perda Sanguínea Muito Recente Anemia Hipocrômica Microcítica
Perda sangüínea crônica perda de FERRO é maior que a sua absorção e reserva hemácias são produzidas com pouca hemoglobina

22
Anemia Ferropriva Decorrente da redução de depósitos de ferro
Sinais: sonolência, fadiga, tonturas, zumbidos, déficit de aprendizagem,palidez cutânea, unhas quebradiças.... Importante: verificar a causa da deficiência de ferro. Tratamento: Aumentar a ingesta( carne, feijão, fígado e vegetais verdes).
.")
23
Carência de Ferro Anemia Ferropênica
Causas: dieta carente, dieta láctea, verminoses, perdas hemorrágicas, gestações repetidas,úlcera gástrica, CA de cólon, doações frequentes de sangue, perda menstrual. Deficiência na Síntese de Hb

24
Carência de Ferro Anemia Ferropênica
ferro porfirina HEME GLOBINA Hemoglobina

25
Anemia megaloblástica
A anemia megaloblástica, também chamada anemia perniciosa, é uma doença na qual a medula óssea produz hemácias (glóbulos vermelhos) e neutrófilos (glóbulos brancos) gigantes e imaturos. Esse distúrbio é provocado pela carência de vitamina B12 ou de ácido fólico no organismo. Uma vez que esses dois fatores são importantes para a síntese de DNA e responsáveis pela eritropoiese, a sua falta causa um defeito na síntese de DNA, levando ao desequilíbrio no crescimento e divisão celular. Alimentos ricos em ácido fólico: frutas, vegetais e cereais. Absorção: no jejuno.
 e neutrófilos (glóbulos brancos) gigantes e imaturos. Esse distúrbio é provocado pela carência de vitamina B12 ou de ácido fólico no organismo. Uma vez que esses dois fatores são importantes para a síntese de DNA e responsáveis pela eritropoiese, a sua falta causa um defeito na síntese de DNA, levando ao desequilíbrio no crescimento e divisão celular. Alimentos ricos em ácido fólico: frutas, vegetais e cereais. Absorção: no jejuno.")
26
Anemia Megaloblástica
Por deficiência de Vit B12 ou de Ácido Fólico. São distúrbios provocados pela síntese comprometida do DNA. A divisão celular é lenta, porém o desenvolvimento citoplasmático progride normalmente, de modo que células megaloblásticas tendem a ser grandes. Transportam O2, porém são mais frágeis

27
CAUSAS SINAIS E SINTOMAS
Atrofia da mucosa gástrica Deficiência de fator intrínseco Auto-agressão contra células parietais Dieta vegetariana estrita Gastrectomia SINAIS E SINTOMAS Palidez cutânea, icterícia discreta, cabelos grisalhos prematuramente, anorexia, sonolência, confusão mental

28
Carência de Vitamina B12 Anemia Perniciosa
Vitamina B proliferação dos glóbulos do sangue integridade das células nervosas Causa: gastrite atrófica, absorção inadequada glossite dormências e falta de sensibilidade nas extremidades deterioração mental irreversível Tratamento: Ao longo de toda a vida- Vitamina B12 (Cianocobalamina)-I.M. na ! Semana- 24/24 horas e depois injeções mensais. Prognóstico: As alterações hematológicas revertem com o tratamento,atingindo valores normais. Se não fizer o tratamento no tempo certo, pode haver sequelas no SNC
-I.M. na ! Semana- 24/24 horas e depois injeções mensais. Prognóstico: As alterações hematológicas revertem com o tratamento,atingindo valores normais. Se não fizer o tratamento no tempo certo, pode haver sequelas no SNC.")
29
Anemia das doenças crônicas
É o seqüestro de Ferro nas células da medula óssea, baço e fígado, devido a patogenias: infecções, neoplasias, cirurgias ou traumas extensos e inflamações no trato digestivo.

30
Anemias por Defeitos Genéticos
Anemia de células falciformes Talassemias Deficiência de glicose-6P desidrogenase (favismo)
")
31
Troca de um aa polar (GLU) por um apolar (VAL) na cadeia .
Anemia Falciforme Troca de um aa polar (GLU) por um apolar (VAL) na cadeia . Hb S A hemoglobina em baixa quantidade de oxigênio se precipita em cristais longos dentro da hemácia FORMA DE FOICE
 por um apolar (VAL) na cadeia . Hb S. A hemoglobina em baixa quantidade de oxigênio se precipita em cristais longos dentro da hemácia. FORMA DE. FOICE.")
32
Anemia Falciforme Essa forma é menos solúvel e danifica a membrana celular célula mais frágil hemólise Ocorre principalmente na população negra (0,3-1%) Durante toda a vida, crises de dor e susceptibilidade aumentada para infecções reduzida tendência a contrair Malária 1. Crise de Dor 2. Icterícia 3. Síndrome Mão-pé 4. Infecções 5. Úlcera de Perna Teste do pezinho Evitar frio e calor extremos
 Durante toda a vida, crises de dor e susceptibilidade aumentada para infecções. reduzida tendência a contrair Malária. 1. Crise de Dor. 2. Icterícia. 3. Síndrome Mão-pé. 4. Infecções. 5. Úlcera de Perna. Teste do pezinho. Evitar frio e calor extremos.")
33
Resistência à Malária Mecanismos de resistência:
Célula com saliências aderência ao endotélio menor quantidade de oxigênio afoiçamento perfuração das membranas parasita depleção do K+ celular não multiplicação dos parasitas; Destruição prematura dos eritrócitos + nutrição deficiente não sobrevivência dos parasitas.

34
Manifestações Clínicas
Iniciam após a 10ª semana de vida, pois o RN é protegido pelos elevados níveis de HbF. Paciente é anêmico, porém assintomático a maior parte do tempo. Bem-estar é interrompido periodicamente por crises. Crises podem ser vaso-oclusivas (dolorosas), aplásicas e megaloblásticas, de seqüestração, e hemolíticas.
, aplásicas e megaloblásticas, de seqüestração, e hemolíticas.")
35
Manifestações Clínicas
Crises vaso-oclusivas mais comum e característica da AF. É causada pela obstrução dos vasos sangüíneos por hemácias falciformes. Ocorre hipóxia tecidual que ocasiona a morte tecidual e dor localizada. É diagnóstico de exclusão. Localização preferencial: ossos, tórax e abdome. Infartos de repetição no baço levam à sua fibrose e atrofia (auto-esplenectomia). Infarto de vasos cerebrais resultando em AVC é a complicação vaso-oclusiva mais grave.
. Infarto de vasos cerebrais resultando em AVC é a complicação vaso-oclusiva mais grave.")
36
Manifestações Clínicas
Crise de Seqüestração são caracterizadas pelo aprisionamento de eritrócitos, especialmente no baço. exacerbação aguda da anemia, reticulocitose persistente, baço hipersensível e de tamanho aumentado e, por vezes, hipovolemia. Ocorreu em 30% das crianças durante 10 anos de seguimento, sendo 15% delas fatais. Recidiva em 50% dos casos, sendo indicada esplenectomia.

37
Deficiência na Síntese de Hb
Talassemia ferro porfirina HEME GLOBINA Hemoglobina Deficiência na Síntese de Hb

38
Talassemia Síntese desequilibrada de globina
quantidades menores de Hb funcionante Talassemia (defeito na formação das cadeias ) Talassemia (defeito na formação das cadeias ) elevada prevalência nos povos do mediterrâneo, sendo a doença genética mais comum em todo o mundo. Talassemias Menores (apenas um gene):discreta anemia Talassemias Maiores (dois genes): anemia severa, icterícia, deformidades ósseas e esplenomegalia.
 Talassemia (defeito na formação das cadeias ) elevada prevalência nos povos do mediterrâneo, sendo a doença genética mais comum em todo o mundo. Talassemias Menores (apenas um gene):discreta anemia. Talassemias Maiores (dois genes): anemia severa, icterícia, deformidades ósseas e esplenomegalia.")
39
Aparece após 3-6 meses de idade
Sinais: Palidez, inapetência, úlceras nas pernas, esplenomegalia, crescimento deficiente. Talassemia maior: morrem precocemente Cuidados: Menor: não necessita de tratamento Maior: manter o nível de hemoglobina, com transfusão de sangue de 3 a 5 semanas. Esplenectomia- pacientes maior que 6 anos- risco de infecção Transplante de medula óssea

40
Favismo Deficiência de glicose-6-fosfato-desidrogenase
Enzima Marcapasso do Ciclo das Pentoses Produção de NadPH (poder redutor) Mantém a Glutationa reduzida Detoxificação do Peróxido de Hidrogênio Carência hemácias sensíveis à oxidação Hemólise
 Mantém a Glutationa reduzida. Detoxificação do Peróxido de. Hidrogênio. Carência hemácias sensíveis à oxidação. Hemólise.")
41
Anemias Decorrentes de Doenças da Medula Óssea
Anemia Aplástica: falta de medula óssea funcionante: lesão das células primitivas da eritropoese. Pode ter como causa o uso de drogas, a hepatite viral e a exposição à radiação ionizante. Leucemias e Tumores na Medula

42
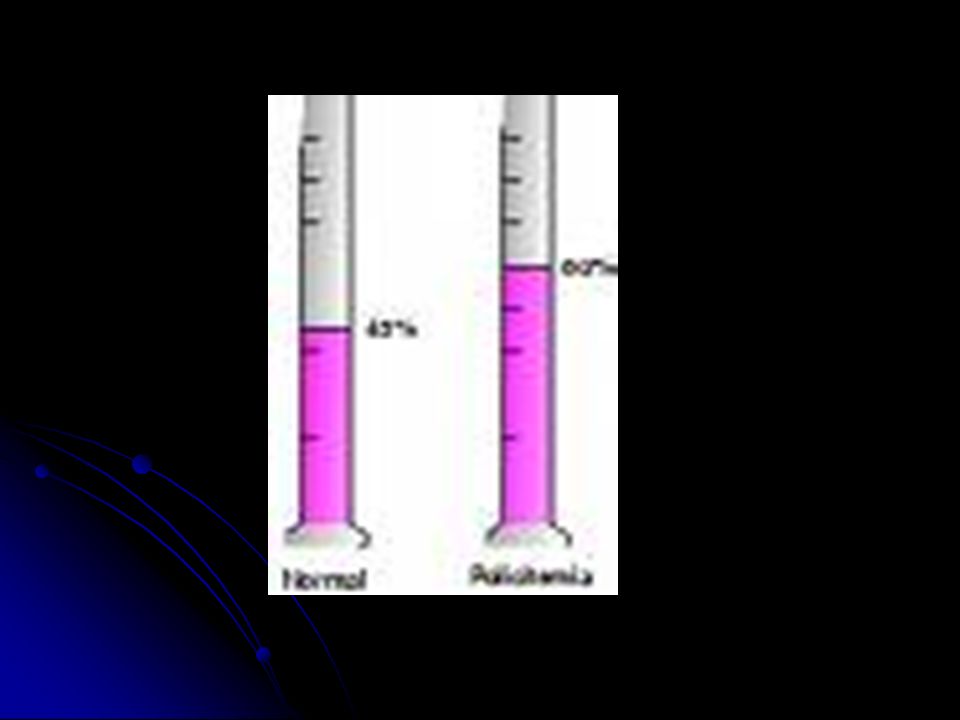
Policitemia A policitemia vera é um aumento anormal das células sangüíneas (principalmente de glóbulos vermelhos), como resultado de um aumento da produção pela medula óssea. A policitemia vera é rara (5 indivíduos:1 milhão). Ocorre com maior freqüência em homens e quase nunca em pessoas com menos de 40 anos. A doença se desenvolve lentamente, em geral após os 50 ou 60 anos de idade e pode progredir até converter-se em uma leucemia mielocítica aguda. A viscosidade do sangue e o aumento de plaquetas resultam em um alto potencial para a formação de coágulos, que pode causar derrame ou um ataque cardíaco. Em alguns pacientes ocorre uma hemorragia porque, apesar da presença de um grande número de plaquetas responsáveis pela coagulação sangüínea, sua capacidade de coagulação está deteriorada. A incidência é maior nas pessoas de ascendência judaica.
, como resultado de um aumento da produção pela medula óssea. A policitemia vera é rara (5 indivíduos:1 milhão). Ocorre com maior freqüência em homens e quase nunca em pessoas com menos de 40 anos. A doença se desenvolve lentamente, em geral após os 50 ou 60 anos de idade e pode progredir até converter-se em uma leucemia mielocítica aguda. A viscosidade do sangue e o aumento de plaquetas resultam em um alto potencial para a formação de coágulos, que pode causar derrame ou um ataque cardíaco. Em alguns pacientes ocorre uma hemorragia porque, apesar da presença de um grande número de plaquetas responsáveis pela coagulação sangüínea, sua capacidade de coagulação está deteriorada. A incidência é maior nas pessoas de ascendência judaica.")
43
Sintomas: fraqueza, fadiga, cefaléia, tontura e falta de ar.
A visão pode ser distorcida e o indivíduo pode apresentar manchas cegas ou pode ver flashes de luz. O sangramento gengival e através de pequenos cortes é comum. A pele, especialmente a da face, pode tornar-se avermelhada. O indivíduo pode sentir um prurido generalizado, sobretudo após um banho quente. Com a evolução do distúrbio, o fígado e o baço podem aumentar de tamanho, causando uma dor maçante e intermitente no abdômen.

44
Prognóstico e tratamento:
Sem tratamento, aproximadamente metade dos indivíduos com policitemia vera sintomáticos morre em menos de 2 anos. Com tratamento, eles vivem, em média, 15 a 20 anos. Normalmente, é realizada a remoção do sangue do corpo através de um procediemnto denominado flebotomia. São removidos 500 ml de sangue em dias alternados até o hematócrito começar a diminuir. Quando ele atinge o nível normal, o sangue é removido em intervalos de alguns meses, de acordo com a necessidade. Esses indivíduos necessitam de quimioterapia para suprimir a produção de células sangüíneas. Comumente, é utilizada a hidroxiuréia, uma droga antineoplásica. Outros medicamentos podem ajudar a controlar alguns dos sintomas. Por exemplo, os anti-histamínicos podem ajudar a aliviar o prurido e a aspirina pode aliviar as sensações de queimação nas mãos e nos pés e também as dores ósseas.

46
LEUCEMIA Leucemia é o câncer dos glóbulos brancos que são produzidos na medula óssea. Por alguma razão, algo acontece de errado (uma mutação) que o organismo não consegue corrigir e a célula alterada, chamada de blasto, começa a se multiplicar dentro da medula óssea substituindo o tecido normal que produz sangue e elementos para coagulação. Estes blastos começam a sair para a circulação sangüínea, onde são detectados. O exame inicial para sua detecção é o hemograma completo.
 que o organismo não consegue corrigir e a célula alterada, chamada de blasto, começa a se multiplicar dentro da medula óssea substituindo o tecido normal que produz sangue e elementos para coagulação. Estes blastos começam a sair para a circulação sangüínea, onde são detectados. O exame inicial para sua detecção é o hemograma completo.")
47
Leucemias As leucemias englobam as formas de câncer que afetam os órgãos que produzem as células do sangue, tanto o sistema linfático como a medula óssea. As leucemias habitualmente afetam os glóbulos brancos. A leucemia pode ser aguda (aparecendo “de repente”) ou crônica (durando mais tempo). A leucemia crônica raramente afeta as crianças, enquanto a leucemia aguda afeta adultos e crianças. A causa da maioria dos tipos de leucemia ainda é desconhecida. A exposição à radiação e a certas substâncias químicas, como o benzeno, e o uso de alguns medicamentos anti-cancer aumentam o risco de leucemia.
 ou crônica (durando mais tempo). A leucemia crônica raramente afeta as crianças, enquanto a leucemia aguda afeta adultos e crianças. A causa da maioria dos tipos de leucemia ainda é desconhecida. A exposição à radiação e a certas substâncias químicas, como o benzeno, e o uso de alguns medicamentos anti-cancer aumentam o risco de leucemia.")
48
Incidência: As leucemias são os cânceres infantis mais comuns, representando 30-35% do total de doenças malignas, sendo que a LLA representa por volta de 75% dos casos de leucemia e o pico de incidência ocorre aos 3-4 anos. Ocorre em proporção um pouco maior nos meninos em relação as meninas. A LLA pode ocorrer em maior freqüência em pacientes portadores de outras doenças, como distúrbios genéticos (síndrome de Down é o exemplo mais comum) ou com imunodeficiência, mas na grande maioria dos casos não há uma explicação causal possível.
 ou com imunodeficiência, mas na grande maioria dos casos não há uma explicação causal possível.")
49
As leucemias representam aproximadamente 2 por cento de todos os cânceres.
Elas atingem mais ou menos 9 em cada pessoas no mundo inteiro a cada ano. Incide mais em homens que em mulheres. A raça branca é mais afetada que outros grupos raciais ou étnicos. Os adultos têm 10 vezes mais chance de desenvolver a leucemia que as crianças. A leucemia aparece mais freqüentemente nos idosos. Quando a leucemia ocorre na criança, ela aparece em geral antes dos 4 anos de idade.

50
Existem quatro tipos principais de leucemia, denominados em função da velocidade de progressão e do tipo de glóbulo branco que afetam. As leucemias linfáticas afetam os linfócitos; as leucemias mielóides (mielocíticas) afetam os mielócitos. Os mielócitos transformam-se em granulócitos. Principais tipos de leucemia Tipo Evolução Glóbulos brancos afectados Leucemia linfocítica aguda (linfoblástica). Rápida Linfócitos Leucemia mielóide aguda (mielocítica,mielogénea, mieloblástica, mielomonocítica). Mielócitos Leucemia linfocítica crónica, incluindo a síndroma de Sézary e a tricoleucemia. Lenta Leucemia mielóide crónica (mielocítica, mielogénea, granulocítica).
. Rápida. Linfócitos. Leucemia mielóide aguda (mielocítica,mielogénea, mieloblástica, mielomonocítica). Mielócitos. Leucemia linfocítica crónica, incluindo a síndroma de Sézary e a tricoleucemia. Lenta. Leucemia mielóide crónica (mielocítica, mielogénea, granulocítica).")
51
Leucemia Aguda Na leucemia aguda as células imaturas do sangue se reproduzem rapidamente, na medula óssea, e impedem as células saudáveis de ganhar a corrente sanguínea. As células imaturas e anormais podem se espalhar para outros órgãos causando lesões. Os dois tipos principais de leucemia aguda envolvem diferentes tipos de células do sangue: Leucemia Linfóide Aguda (ou leucemia linfocítica aguda ou LLA): É o tipo mais comum de leucemia que afeta as crianças, primariamente aquelas antes dos 10 anos de idade. Os adultos, algumas vezes, podem desenvolver a LLA, mas ela é rara em pessoas acima dos 50 anos. A LLA ocorre quando células primitivas do sangue chamadas linfoblastos (linfócitos em desenvolvimento) não amadurecem e se multiplicam em excesso. Estas células anormais impedem que as células normais sejam liberadas. Elas podem se acumular nos gânglios linfáticos (linfonodos) e causar inchaço (ínguas).
: É o tipo mais comum de leucemia que afeta as crianças, primariamente aquelas antes dos 10 anos de idade. Os adultos, algumas vezes, podem desenvolver a LLA, mas ela é rara em pessoas acima dos 50 anos. A LLA ocorre quando células primitivas do sangue chamadas linfoblastos (linfócitos em desenvolvimento) não amadurecem e se multiplicam em excesso. Estas células anormais impedem que as células normais sejam liberadas. Elas podem se acumular nos gânglios linfáticos (linfonodos) e causar inchaço (ínguas).")
52
Irmãos de crianças com LLA têm um risco de 2 a 4 vezes maior de também apresentarem LLA em relação à população geral e este risco ainda se multiplica nos gêmeos idênticos. É uma doença fulminante que, na ausência de intervenção médica, provoca a morte dentro de poucos meses. As Leucemias Linfóides Crônicas (LLC) não se manifestam na faixa pediátrica.
 não se manifestam na faixa pediátrica..")
53
Sintomas Dois terços das crianças com LLA mostram sinais e sintomas da doença num período de um mês até o diagnóstico. Os primeiros sintomas não são específicos e incluem falta de apetite, irritabilidade e fraqueza. Com a progressão da doença na medula óssea, aparecem palidez, sangramentos não ligados à traumas e febre. Em 25% dos portadores de LLA ao diagnóstico, há dores ósseas e nas juntas devido à doença estar aumentando a pressão dentro dos ossos.

54
TRATAMENTO Os tratamentos podem diferir um pouco levando em conta a idade do paciente e características dos exames de sangue e medula óssea e exames radiológicos. O programa geral de tratamento inclui a indução, onde é administrado a quimioterapia até que a medula óssea não mostre mais células cancerosas e tratamento profilático do sistema nervoso central. Depois é feito a consolidação e a manutenção, tudo com quimioterapia sistêmica durante um longo tempo (mais de 2 anos) para não deixar nenhuma célula maligna escapar. Os melhores centros de oncologia infantil do mundo curam a LLA numa taxa de até 70%, onde os doentes ficam mais de 5 anos livres da doença.
 para não deixar nenhuma célula maligna escapar. Os melhores centros de oncologia infantil do mundo curam a LLA numa taxa de até 70%, onde os doentes ficam mais de 5 anos livres da doença.")
55
Leucemia Mielóide Aguda (LMA): Representa aproximadamente 50 por cento das leucemias diagnosticadas em adolescentes e em pessoas por volta dos 20 anos de idade. É a forma mais comum de leucemia aguda em adultos. Ela ocorre quando células primitivas do sangue chamadas mieloblastos reproduzem sem se desenvolver em células normais do sangue. Os mieloblastos imaturos se acumulam na medula óssea e interferem com a produção das células normais do sangue. Isto leva a anemia (quantidade insuficiente de células vermelhas no sangue) e infecções freqüentes porque não há leucócitos (células brancas) suficientes no sangue.
 e infecções freqüentes porque não há leucócitos (células brancas) suficientes no sangue.")
56
Leucemia Crônica Leucemia Linfóide Crônica (LLC ou leucemia linfocítica crônica): É rara em pessoas abaixo dos 30 anos de idade. Ela é mais freqüente a medida que a pessoa envelhece. O maior número dos casos ocorre em pessoas em torno dos 60 e 70 anos de idade. Leucemia Mielóide Crônica (LMC): Ocorre em pessoas entre os 25 e 60 anos de idade. Nesta forma de leucemia, o tipo de célula anormal do sangue é chamada célula mielóide.
: É rara em pessoas abaixo dos 30 anos de idade. Ela é mais freqüente a medida que a pessoa envelhece. O maior número dos casos ocorre em pessoas em torno dos 60 e 70 anos de idade. Leucemia Mielóide Crônica (LMC): Ocorre em pessoas entre os 25 e 60 anos de idade. Nesta forma de leucemia, o tipo de célula anormal do sangue é chamada célula mielóide.")
57
Quadro Clínico SÍNDROME ANÊMICA: Caracteriza-se por uma anemia crescente, acompanhada de palidez da pele e das mucosas, falta de ar que aumenta com o esforço físico, cansaço, dores de cabeça, agitação ou sonolência e taquicardia SÍNDROME HEMORRÁGICA: Decorrente da progressiva diminuição ou ausência das plaquetas do sangue. SÍNDROME FEBRIL: Mostra-se como uma febre de origem indeterminada, muitas vezes com tosse, dor abdominal, dor de garganta, fazendo o paciente procurar o médico. SÍNDROME TUMORAL: Ocorre pelo acúmulo de células sangüíneas imaturas em órgãos como o fígado, baço, gânglios línfáticos e genitais. Pode haver comprometimento dos ossos e articulações, em até 30% dos casos, causando dor de forte intensidade. Quando atinge o sistema nervoso central é acompanhada de vômitos, dores de cabeça, diminuição de sensibilidade ou de força motora. Os testículos podem ser afetados em até 20% dos casos, tornando-se inchados, mas a dor não costuma ser intensa. Os ovários, em geral, não costumam ser comprometidos.

58
Transplante de medula óssea (TMO)
O transplante de medula óssea (TMO) é modalidade terapêutica utilizada no tratamento de inúmeras das doenças hematológicas, benignas ou malignas; hereditárias ou adquiridas. O fundamento lógico para o transplante de células progenitoras está baseado no fato de que todas as células maduras que circulam no sangue: glóbulos vermelhos, glóbulos brancos e plaquetas provém de uma única célula, contida na medula óssea, denominada célula progenitora ou “stem cell”, e atualmente o termo mais amplamente aceito é células progenitoras hematopoéticas (TCPH).
 é modalidade terapêutica utilizada no tratamento de inúmeras das doenças hematológicas, benignas ou malignas; hereditárias ou adquiridas. O fundamento lógico para o transplante de células progenitoras está baseado no fato de que todas as células maduras que circulam no sangue: glóbulos vermelhos, glóbulos brancos e plaquetas provém de uma única célula, contida na medula óssea, denominada célula progenitora ou stem cell , e atualmente o termo mais amplamente aceito é células progenitoras hematopoéticas (TCPH).")
59
A realização do transplante consiste na retirada de células progenitoras hematopoéticas. Tais células estão localizadas em adultos principalmente nos ossos chatos como a bacia, esterno, costela e vértebras. As células progenitoras infundidas na corrente sangüínea se implantam na medula óssea iniciando a reconstituição hematopoética do paciente, após regime de condicionamento. O condicionamento é o uso de altas doses de quimioterapia associados ou não à radioterapia corporal para que o paciente seja tratado de sua doença hematológica. Com a infusão de células progenitoras suficientes do paciente (denominado transplante autólogo) ou de um doador próximo e compatível (denominado transplante alogênico), a função da medula e a produção das células do sangue são restauradas de maneira suficiente a permitir a recuperação de um tratamento intensivo.
 ou de um doador próximo e compatível (denominado transplante alogênico), a função da medula e a produção das células do sangue são restauradas de maneira suficiente a permitir a recuperação de um tratamento intensivo.")
60
Há três formas de transplante: 1 - Alogênico: as células progenitoras provém de um doador previamente selecionado por testes de compatibilidade, principalmente o HLA (antígeno de hispocompatibilidade leucocitária) normalmente identificado entre os familiares ou em bancos de medula óssea. Os bancos de medula óssea podem ter cadastrados doadores adultos ou bancos de cordão umbilical Autólogo: as células progenitoras provém do próprio paciente Singênico: as células progenitoras provém de gêmeos idênticos (univitelinos).
..")
61
PARA O DOADOR: A doação de medula óssea classicamente é feita por meio de um procedimento, de aproximadamente 90 minutos, em que são realizadas múltiplas punções com agulhas, nos ossos posteriores da bacia sendo retirado o produto através de aspiração. Retira-se um volume de medula do doador de, no máximo, 10% do seu peso, de 10 a 15 ml/kg de peso do receptor. Esta retirada não causa qualquer comprometimento à saúde do doador. PARA O PACIENTE Depois de se submeter a um tratamento que reduz drasticamente a produção normal de sangue, o paciente recebe as células progenitoras transfundidas para a corrente sangüínea. As células progenitoras, uma vez na corrente sangüínea, circulam e vão se alojar na medula óssea e voltam a se proliferar. Durante o período em que estas células ainda não são capazes de produzir glóbulos brancos, vermelhos e plaquetas em quantidade suficiente para manter as taxas dentro da normalidade, o paciente fica mais exposto a episódios infecciosos e ou hemorragias. Por esta razão, deve ser mantido preferencialmente internado e em regime de isolamento. Cuidados com a dieta e higiene são necessários.

64
HEMOFILIA A hemofilia é um distúrbio hemorrágico hereditário, decorrentes da deficiência de fator VIII (hemofilia A) ou IX (hemofilia B), caracterizada por um defeito na coagulação. O sangue é feito por várias substâncias, onde cada uma tem uma função. Algumas delas são as proteínas chamadas fatores da coagulação, que ajudam a estancar as hemorragias. Esses fatores são numerados, em algarismos romanos (I a XIII) e trabalham como uma equipe, onde cada um tem seu momento de ação, passando instruções ao seguinte. Por isso, o sangue da pessoa com hemofilia demora mais para formar um coágulo e, quando este se forma, não é capaz de fazer o sangue parar de escorrer pelo local da lesão.
 ou IX (hemofilia B), caracterizada por um defeito na coagulação. O sangue é feito por várias substâncias, onde cada uma tem uma função. Algumas delas são as proteínas chamadas fatores da coagulação, que ajudam a estancar as hemorragias. Esses fatores são numerados, em algarismos romanos (I a XIII) e trabalham como uma equipe, onde cada um tem seu momento de ação, passando instruções ao seguinte. Por isso, o sangue da pessoa com hemofilia demora mais para formar um coágulo e, quando este se forma, não é capaz de fazer o sangue parar de escorrer pelo local da lesão.")
65
SINTOMAS Sangramentos, principalmente dentro das juntas e dos músculos. Apresentam hemorragias espontâneas, ou seja, repentinas e sem causa aparente. As simples atividades normais da vida diária como caminhar e correr podem produzir hemorragias. As hemorragias espontâneas geralmente acontecem nas partes do corpo onde há muita atividade e esforço, principalmente nas juntas (articulações). É importante lembrar que quando uma pessoa com hemofilia se machuca, não sangra mais rápido do que uma outra sem hemofilia, apenas fica sangrando durante um tempo maior e pode recomeçar a sangrar vários dias depois de um ferimento ou de uma cirurgia.
. É importante lembrar que quando uma pessoa com hemofilia se machuca, não sangra mais rápido do que uma outra sem hemofilia, apenas fica sangrando durante um tempo maior e pode recomeçar a sangrar vários dias depois de um ferimento ou de uma cirurgia.")
66
Tratamento Evitar o uso de AAS e outros que interferem na coagulação
Assistência dentária Infusão de fatores de coagulação- pode desenvolver inibidores Evolução: Hemartroses resultam em deformações. Doença leve: vida normal

Apresentações semelhantes