Carregar apresentação
A apresentação está carregando. Por favor, espere

2
Miguel www. cromatina. icb. ufmg. br

3
Seqüência e qualidade

4
Uso da bioinformática na análise genômica

5
iniciadores crescem até que uma unidade interruptora entre
ATCTCGTAGCT ATCTCGTAGCTA A ATCTCGTAGCTAG G ATCTCGTAGCTAGC C ATCTCGTAGCTAGCT T ATCTCGTAGCTAGCTA A ATCTCGTAGCTAGCTAC C ATCTCGTAGCTAGCTACG G ATCTCGTAGCTAGCTACGA A ATCTCGTAGCTAGCTACGAC C ATCTCGTAGCTAGCTACGACG G ATCTCGTAGCTAGCTACGACGT T ATCTCGTAGCTAGCTACGACGTC C ATCTCGTAGCTAGCTACGACGTCT T ATCTCGTAGCTAGCTACGACGTCTA A TAGAGCATCGATCGATGCTGCAGATGATGCTAGCATCGGCTAGGCGACG iniciadores crescem até que uma unidade interruptora entre uma fita molde é sequenciada de cada vez eletroforese capilar e leitura da fluorescência da unidade interruptora

6
Receber Processar Anotar Depositar
Início Bioinformática Receber Processar Anotar Depositar Fim

7
Processamento de seqüências
20 30 10 cromatograma acgatctcgctagctgctactgtagccgcgattattcgcgatctacgtatatcgcgatcgatc 10 20 30 40 50 O programa Phred lê o cromatograma e nomeia as bases Cada base tem uma chance de erro de sua nomeação (10% = 0,1) A escala de Phred é semelhante à de pH multiplicado por 10: - chance de erro de 0,001 = 10-3 = Phred 30 A nomeação é praticamente aleatória no início e no final, onde a chance de erro é alta (baixo valor de Phred)
 A escala de Phred é semelhante à de pH multiplicado por 10: - chance de erro de 0,001 = 10-3 = Phred 30. A nomeação é praticamente aleatória no início e no final, onde a chance de erro é alta (baixo valor de Phred)")
8
I Brazilian Workshop on Bioinformatics October 18th, 2002, Gramado, RS, Brazil

9
Estratégias Genômicas

10
Crescimento do GenBank
Seqüências 15 milhões 24h Europeu Japonês 606 1982 1983 1984 1985 1986 1987 1988 1989 1990 1991 1992 1993 1994 1995 1996 1997 1998 1999 2000 2001 Ano

11
(seqüenciador grande)
Seqüencias do DNA (genoma) Seqüências do mRNA (genes expressos) Amostragem tecidos momentos Eucariotos mRNA Repetição calculada draft = 5x finished = 10x Genoma pequeno (seqüenciador grande) TR cDNA
 Seqüências do mRNA. (genes expressos) Amostragem. tecidos. momentos. Eucariotos. mRNA. Repetição calculada. draft = 5x. finished = 10x. Genoma pequeno. (seqüenciador grande) TR. cDNA.")
12
Seqüenciamento parcial de transcritos

13
EST (Etiqueta de Seqüência Expressa)
Seqüênciamento de genes expressos: Documentar a existência de transcritos gênicos num transcriptoma [otorrin... e ...damonh...] EST (Etiqueta de Seqüência Expressa) seqüenciamento único de cada cDNA extremidades 5’ ou 3’ ORESTES (ESTs ricas em ORFs) seqüenciamento único do amplicon derivado de cDNA por PCR inespecífico prevalece o centro do cDNA (cds)
 seqüenciamento único de cada cDNA. extremidades 5’ ou 3’ ORESTES (ESTs ricas em ORFs) seqüenciamento único do amplicon derivado de cDNA por PCR inespecífico. prevalece o centro do cDNA (cds)")
14
Um mRNA & suas ESTs 5’EST 3’EST 5’EST 3’EST mRNA ATG AUG cDNA (fita +)
ATCATGACTTACGGGCGCGCGAT AAATTTATTATCC (T)18 5’EST cDNA (fita -) 3’EST mRNA AUG cDNA (fita +) (A)200 (A)18 GGCGCGCGATATCC AAATTTATTATCCATCTACG (T)18 5’EST cDNA (fita -) 3’EST
18. 5’EST. cDNA (fita -) 3’EST. mRNA. AUG. cDNA (fita +) (A)200. (A)18. GGCGCGCGATATCC. AAATTTATTATCCATCTACG. (T)18. 5’EST. cDNA (fita -) 3’EST.")
15
PCR inespecífico & seu ORESTES
amplicon (fita -) amplicon (fita +) PCR (60ºC) +ORESTES (outros iniciadores) mRNA amplicon (fita +) AUG GGGCGCGCGATATCGAAAAATTTATAAGGCTAG (A)200 CCCCGGCGGCTCGGCCGGGGAGATCGATCATGAC AGATCGATCATGACTTACGGGCGCGCGATATCG ORESTES cDNA (fita -) Iniciador (60ºC ºC)
 amplicon (fita +) PCR. (60ºC) +ORESTES (outros iniciadores) mRNA. amplicon (fita +) AUG. GGGCGCGCGATATCGAAAAATTTATAAGGCTAG. (A)200. CCCCGGCGGCTCGGCCGGGGAGATCGATCATGAC. AGATCGATCATGACTTACGGGCGCGCGATATCG. ORESTES. cDNA (fita -) Iniciador. (60ºC 37ºC)")
16
250 200 150 100
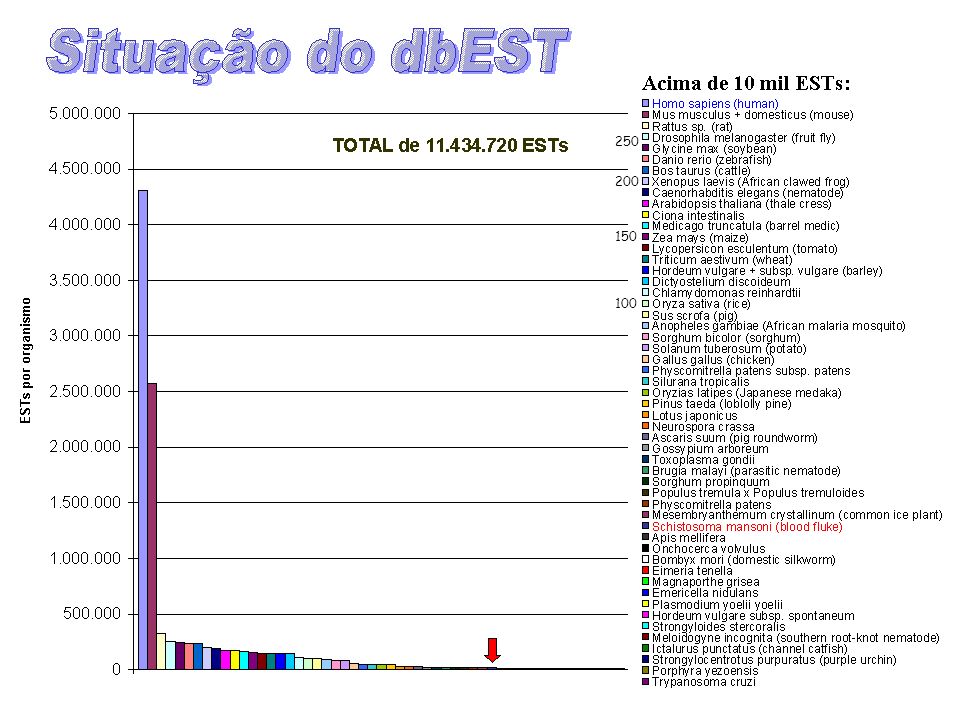
17
Bases de dados

18
O formato FASTA, o mais simples, é anotado
>Gene EST com homologia... ACTATTACGGCGTAGCTGTAGCTACGTAGCTAGCTGATGCTGACTGATCGTAGCTAGCTGACTGATCGTACGTAGTGTTTTTTTACGTGCGTATTtCTagCTaGtc Seqüências > 50 nt, sem ambiguidades e com anotação, ganham entrada no Entrez Protein/Nucleotide

19
Transcriptoma de S. mansoni Rede Genoma de Minas Gerais
Resultados esperados Rede Genoma de Minas Gerais dbEST: ESTs 5’ e 3’ Trace Arquive: dados originais Entrez Nucleotide: > 50 nt, em fase, com anotação Entrez Protein: proteínas deduzidas selecionadas mineração automática (KOG, BioCarta e KEGG) mineração manual (grupos) RefSeq: seqüências de referência UniGene: dados de expressão diferencial (microarray e DGED) número I.M.A.G.E.
 mineração manual (grupos) RefSeq: seqüências de referência. UniGene: dados de expressão diferencial (microarray e DGED) número I.M.A.G.E.")
20
Anotação

21
O mundo

22
Alinhador local Identifica, numa coleção de seqüências, as que apresentam alinhamento com a sua. Fragmenta sua seqüência e procura homologia no banco de dados. Descarta todas as pesquisas com pontuação pequena (score baixo) e vai alinhando a vizinhança das com pontuação boa, até chegar ao máximo valor. É fácil verificar que algumas regiões de certos genes alinham bem, mas outras pouco conservadas, não. O Alinhador Local não quer chegar ao alinhamento final, ele só quer identificar sequências com um nível de homologia significativo
 e vai alinhando a vizinhança das com pontuação boa, até chegar ao máximo valor. É fácil verificar que algumas regiões de certos genes alinham bem, mas outras pouco conservadas, não. O Alinhador Local não quer chegar ao alinhamento final, ele só quer identificar sequências com um nível de homologia significativo.")
23
Alinhamento local O fundamento teórico é que a função gênica está quase sempre confinada em domínios contínuos de uma proteína Se não fosse assim, não teria sentido usar...

24
Programas BLAST & Bancos
Há vários Programas BLAST úteis Alguns são usados quando a sua sequência é de nucleotídeos (BLASTn, BLASTx e tBLASTx) Outros são usados quando a sua seqüência é de aminoácidos (BLASTp) E vários bancos de dados para escolher (nr, pdb, dbEST, yeast, month, etc...) Ou usa-se limites [organism]
 Outros são usados quando a sua seqüência é de aminoácidos (BLASTp) E vários bancos de dados para escolher (nr, pdb, dbEST, yeast, month, etc...) Ou usa-se limites [organism]")
25
BLASTn e BLASTx A EST identifica o gene homólogo: BLASTn
A EST identifica proteína ortóloga de outro organismo - a evolução conservou a proteína enquanto o DNA divergiu: BLASTx BLASTx: a EST traduzida em seis proteínas 1 existe, 5 não... O mundo Blast é assim

26
tBLASTx tBLASTx traduz sua seqüência de nucleotídeos para proteína nas 6 possibilidades, exatamente como BLASTx Depois pesquisa com essas 6 proteínas deduzidas, um banco de dados de nucleotídeos também traduzido dessa maneira Pra que serve? Pois imagine que a telomerase de Euplotes seja parecida com a telomerase humana, mas os dois DNA não! Traduzindo a seqüência pesquisada e o banco de dados dbEST foi possível encontrar seqüências da telomerase humana

27
bioinformática Receber Processar Anotar Depositar

28
Anotação Reversa

29
3. Quais proteínas vamos estudar?

30
Anotação Reversa Proteína pré-selecionada EST
Algoritmo de seleção de clones com CDS completo Patrimônio de um projeto EST Proteína pré-selecionada EST Saldo de códons positivo Anotação Reversa

31
Demonstração de agente selecionador de clones
(utilizando 16 mil ESTs dbEST) Identificador da via COG com presença de genes eucarióticos (Dm e Ce) Número de proteínas depositadas (azul) e de ESTs com saldo de códons positivo (vermelho)
 Identificador da via COG com presença de genes eucarióticos (Dm e Ce) Número de proteínas depositadas (azul) e de. ESTs com saldo de códons positivo (vermelho)")
33
um aglomerado = um gene

34
Aglomerados ou Clusters
Uma das atividades em bioinformática é formar aglomerados de todas as sequências geradas no projeto (as figurinhas de um álbum) Podemos saber quantas vezes um gene foi seqüenciado, e detectar os freqüentes! E quantos dos genes foram detectados Usa-se também para validar bibliotecas
 Podemos saber quantas vezes um gene foi seqüenciado, e detectar os freqüentes! E quantos dos genes foram detectados. Usa-se também para validar bibliotecas.")
35
Programas para aglomerar
Icatools Phrap Cap3, Cap4 Swat BLAST MegaBLAST Um aglomerado = Um gene

36
Qualidade das bibliotecas (100 primeiras ESTs)
Número de seqüências Boa biblioteca ? 1 2 3 4 5 7 9 11 Freqüência em que uma EST foi amostrada

37
UniGene Organização das sequências do GenBank em um conjunto de aglomerados Cada aglomerado do UniGene contém as sequências que representam um gene único E também informações relacionadas, como em que tecidos o gene é expresso, etc. E também onde está mapeado

38
MegaBLAST gera o UniGene
Todas ESTs contra todas Detecção de homologia > 96% de identidade > 70% do potencial Aglomerar

39
Resultado parcial com 14.853 seqüências
12/03/2003 Seqüências boas UFMG UFOP Produção por laboratório

40
Construção de UniGene para AW1
(5.145 ESTs correspondem a clusters) Etapa Número de seqüências no aglomerado Número de algomerados identidade > 96 % alinhamento > 70 % do potencial
 Etapa. Número de seqüências no aglomerado. Número de algomerados. identidade > 96 % alinhamento > 70 % do potencial.")
41
Outros serviços online

42
Interface gráfica Alternativa para encontrar só o gene

43
Online Mendelian Inheritance in Man
Um catálogo de genes humanos e anomalias genéticas de autoria do Dr. Victor A. McKusick e seus colaboradores e desenvolvido para a Web pelo NCBI Funciona como uma revisão já feita

44
SNP catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctaactagctgactg
catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatggtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctatctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgattgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg catcgatcgatcgtcgtagctacgtagctagctagctagctagctagctagctagctgactg SNP

45
No NCBI é acessado um banco de dados: MMDB
Molecular Modelling DataBase (PDB sem teóricos) O banco de dados PDB tem um mirror no Brasil Arquivos do tipo “1MEY.pdb” são descarregados As coordenadas 3D de totos os átomos As proteínas podem ser vistas com programas (RasMol) ou direto no navegador (Plug-in Chime)
 O banco de dados PDB tem um mirror no Brasil. Arquivos do tipo 1MEY.pdb são descarregados. As coordenadas 3D de totos os átomos. As proteínas podem ser vistas com programas (RasMol) ou direto no navegador (Plug-in Chime)")
46
Modelagem Molecular por Homologia
A proteína precisa ter uma ortóloga no PDB Pode ser automaticamente modelada pelo Swiss Model (Modeller na UFMG) Já modelaram todas proteínas confira 3DCrunch:
 Já modelaram todas proteínas. confira 3DCrunch:")
47
Rede Genoma de Minas Gerais

48
Arquitetura computacional
(estações de trabalho e instalações CENAPAD/MG-CO) metionina.cenapad.ufmg.br Processamento, Linux RedHat: Phred, BLAST, Java 2x Xeon 2,4 GHz, 4 GB RAM, 2x 36 GB HD, RAID 0 adenina.cenapad.ufmg.br Armazenamento de dados, Linux RedHat: Oracle 2x PIII 1,4 GHz, 4GB RAM, 3x 36 GB HD, RAID 5 Unidade de fita DAT 20/40 bionfo.cenapad.ufmg.br Interface Web, Windows 2000: QuickPlace 1x PIII 1,4 GHz, 512 MB RAM, 2x 18 GB HD, RAID 1
 metionina.cenapad.ufmg.br. Processamento, Linux RedHat: Phred, BLAST, Java. 2x Xeon 2,4 GHz, 4 GB RAM, 2x 36 GB HD, RAID 0. adenina.cenapad.ufmg.br. Armazenamento de dados, Linux RedHat: Oracle. 2x PIII 1,4 GHz, 4GB RAM, 3x 36 GB HD, RAID 5. Unidade de fita DAT 20/40. bionfo.cenapad.ufmg.br. Interface Web, Windows 2000: QuickPlace. 1x PIII 1,4 GHz, 512 MB RAM, 2x 18 GB HD, RAID 1.")
49
T C A G Osvaldo Carvalho Farah Coordenador de Computação Científica:
LCC-CENAPAD A T G C BIOINFORMÁTICA UFMG Osvaldo Carvalho Farah Coordenador de Computação Científica: Fabiano Peixoto Equipe: Operação Suporte Oracle J. Miguel Ortega Estudantes de Doutorado: Alessandra Faria-Campos Anotação Maurício Mudado* Bases de dados Saulo de Paula* Microarray Cristiane Nobre Redes neurais Daniela Campos Genoma Iniciação Científica: Estevam Bravo-Neto (ciências biológicas)* João Torres (ciências da computação)
* João Torres (ciências da computação)")
Apresentações semelhantes




 dos genes e seu armazenamento>")
















>")