Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Uso da bioinformática na análise genômica

2
ATCTCGTAGCTA ATCTCGTAGCTAGCTACGACGTCTA ATCTCGTAGCTAGCTA ATCTCGTAGCTAG ATCTCGTAGCTAGC ATCTCGTAGCTAGCT ATCTCGTAGCTAGCTAC ATCTCGTAGCTAGCTACG ATCTCGTAGCTAGCTACGA ATCTCGTAGCTAGCTACGAC ATCTCGTAGCTAGCTACGACG ATCTCGTAGCTAGCTACGACGT ATCTCGTAGCTAGCTACGACGTC ATCTCGTAGCTAGCTACGACGTCT ATCTCGTAGCT A G C T A C G A C G T C T A TAGAGCATCGATCGATGCTGCAGATGATGCTAGCATCGGCTAGGCGACG

3
Start End

4
Processamento de seqüências
20 30 10 cromatograma acgatctcgctagctgctactgtagccgcgattattcgcgatctacgtatatcgcgatcgatc O programa Phred lê o cromatograma e nomeia as bases Cada base tem uma chance de erro de sua nomeação (10% = 0,1) A escala de Phred é semelhante à de pH multiplicado por 10: - chance de erro de 0,001 = 10-3 = Phred 30 A nomeação é praticamente aleatória no início e no final, onde a chance de erro é alta (baixo valor de Phred)
 A escala de Phred é semelhante à de pH multiplicado por 10: - chance de erro de 0,001 = 10-3 = Phred 30. A nomeação é praticamente aleatória no início e no final, onde a chance de erro é alta (baixo valor de Phred)")
5
In the Pursuit of Optimal Sequence Trimming Parameters for EST Projects
Fabiano C. Peixoto & J. Miguel Ortega LCC-CENAPAD A T G C BIOINFORMÁTICA UFMG

6
Noticed: BLAST results Phred 15 Too much trimming 10 20 30 40 50

7
.TGAAGCTTTCAGCTTCTTTAGGAGGATCGTTTTTAGAATCCCCTGCAAC
GTTACCACGGTGGATTTCACTGACTGCGACGTTCTTAACGTTGAATCCAA CGttGCTACCAgggagagcctcagtaagtgcttcatgatgcatttcgaca gaattgacttcagtcgacaaaccttgcggagcaaaagtgacgaccatacc aggcttgatgataccagtttcaacgcctcggggccaggctggcgtgaaca gggcctagcgggtccgcgggggaagggtcccggctcaatccaccaataga gcggagctaaagtgacgggggcgcca Phred 15 Query: 469 TTAGGAGGATCGTTTTTAGAATCCCCTGCAACGTTACCACGGTGGATTTCACTGACTGCG 528 |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||| Sbjct: 1038 ttaggaggatcgtttttagaatcccctgcaacgttaccacggtggatttcactgactgcg 979 Query: 529 ACGTTCTTAACGTTGAATCCAACGTTGCTACCAgggagagcctcagtaagtgcttcatga 588 ||||||||||||||||| || |||||||||||||||||| |||||||||||||||||||| Sbjct: 978 acgttcttaacgttgaagcccacgttgctaccagggagaccctcagtaagtgcttcatga 919 Query: 589 tgcatttcgacagaattgacttcagtcgacaaaccttgcggagcaaaagtgacgaccata 648 |||||||||||||| |||||||||| |||| ||||||||||| ||||||||||||||||| Sbjct: 918 tgcatttcgacagacttgacttcagccgaccaaccttgcggaccaaaagtgacgaccata 859 Query: 649 ccaggcttgatgataccagtttcaacgc 676 |||||||||||||||||||||||||||| Sbjct: 858 ccaggcttgatgataccagtttcaacgc 831

8
Experimental approach
Sequences: pUC18 plasmidial vector (published sequence) Sequence reaction: Single pool - 3 plates (96 samples) MegaBACE sequencer 3 reads for each plate, esd processing reads Processing: BLAST (MegaBLAST, as in UniGene) Phred trim: a chromatogram analyzer trim_alt: trim_cutoff parameter 1% up to 25%
 Sequence reaction: Single pool - 3 plates (96 samples) MegaBACE sequencer. 3 reads for each plate, esd processing reads. Processing: BLAST (MegaBLAST, as in UniGene) Phred. trim: a chromatogram analyzer. trim_alt: trim_cutoff parameter 1% up to 25%")
9
Phred

10
16% 17% Trim_alt sequence Additional bases BLAST gaps/missmatches (% of bases) 3%
 3%")
11
Crescimento do GenBank
Seqüências 15 milhões 24h Europeu Japonês 606 1982 1983 1984 1985 1986 1987 1988 1989 1990 1991 1992 1993 1994 1995 1996 1997 1998 1999 2000 2001 Ano

12
(seqüenciador grande) Eucariotos
Seqüencias do DNA (genoma) Seqüências do mRNA (genes expressos) Repetição calculada draft = 5x finished = 10x Amostragem tecidos momentos Genoma pequeno (seqüenciador grande) Eucariotos mRNA TR cDNA
 Seqüências do mRNA. (genes expressos) Repetição calculada. draft = 5x. finished = 10x. Amostragem. tecidos. momentos. Genoma pequeno. (seqüenciador grande) Eucariotos. mRNA. TR. cDNA.")
13
Seqüenciamento parcial de transcritos

14
EST (Etiqueta de Seqüência Expressa)
Seqüênciamento de genes expressos: Documentar a existência de transcritos gênicos num transcriptoma [otorrin... e ...damonh...] EST (Etiqueta de Seqüência Expressa) seqüenciamento único de cada cDNA extremidades 5’ ou 3’ ORESTES (ESTs ricas em ORFs) seqüenciamento único do amplicon derivado de cDNA por PCR inespecífico prevalece o centro do cDNA (cds)
 seqüenciamento único de cada cDNA. extremidades 5’ ou 3’ ORESTES (ESTs ricas em ORFs) seqüenciamento único do amplicon derivado de cDNA por PCR inespecífico. prevalece o centro do cDNA (cds)")
15
Um mRNA & suas ESTs 5’EST 3’EST 5’EST 3’EST mRNA AUG ATG cDNA (fita +)
ATCATGACTTACGGGCGCGCGAT AAATTTATTATCC (T)18 5’EST cDNA (fita -) 3’EST mRNA AUG cDNA (fita +) (A)200 (A)18 GGCGCGCGATATCC AAATTTATTATCCATCTACG (T)18 5’EST cDNA (fita -) 3’EST
18. 5’EST. cDNA (fita -) 3’EST. mRNA. AUG. cDNA (fita +) (A)200. (A)18. GGCGCGCGATATCC. AAATTTATTATCCATCTACG. (T)18. 5’EST. cDNA (fita -) 3’EST.")
16
PCR inespecífico & seu ORESTES
amplicon (fita -) amplicon (fita +) PCR (60ºC) +ORESTES (outros iniciadores) mRNA amplicon (fita +) AUG GGGCGCGCGATATCGAAAAATTTATAAGGCTAG (A)200 CCCCGGCGGCTCGGCCGGGGAGATCGATCATGAC AGATCGATCATGACTTACGGGCGCGCGATATCG ORESTES cDNA (fita -) Iniciador (60ºC ºC)
 amplicon (fita +) PCR. (60ºC) +ORESTES (outros iniciadores) mRNA. amplicon (fita +) AUG. GGGCGCGCGATATCGAAAAATTTATAAGGCTAG. (A)200. CCCCGGCGGCTCGGCCGGGGAGATCGATCATGAC. AGATCGATCATGACTTACGGGCGCGCGATATCG. ORESTES. cDNA (fita -) Iniciador. (60ºC 37ºC)")
17
250 200 150 100
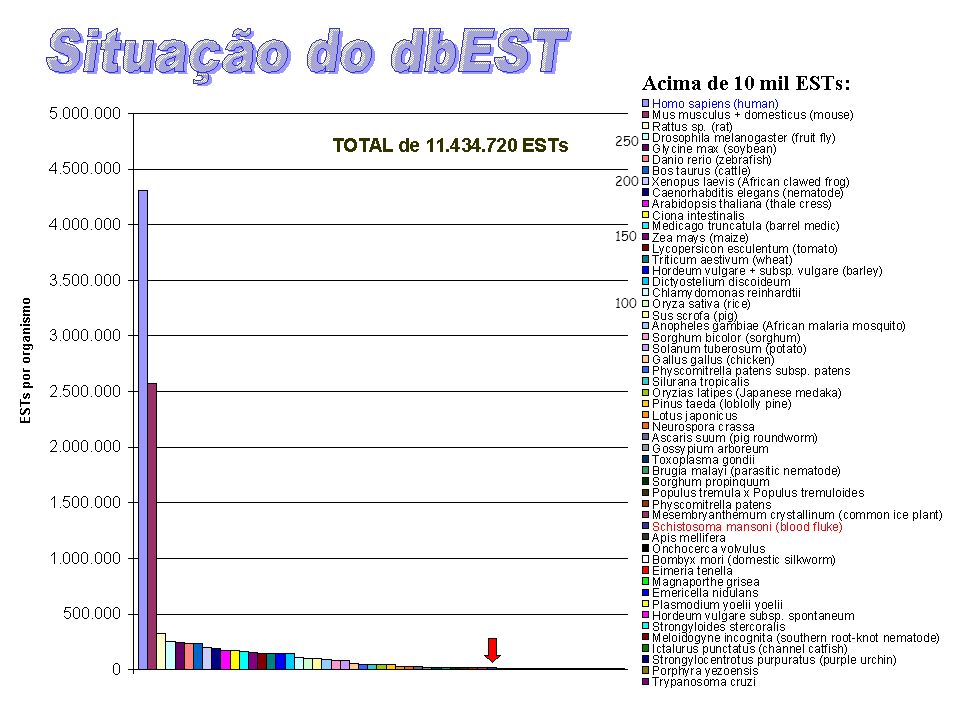
18
O formato FASTA, o mais simples, é anotado
>Gene EST com homologia... ACTATTACGGCGTAGCTGTAGCTACGTAGCTAGCTGATGCTGACTGATCGTAGCTAGCTGACTGATCGTACGTAGTGTTTTTTTACGTGCGTATTtCTagCTaGtc Seqüências > 50 nt, sem ambiguidades e com anotação, ganham entrada no Entrez Protein/Nucleotide

19
O mundo

20
Alinhador local Identifica, numa coleção de seqüências, as que apresentam alinhamento com a sua. Fragmenta sua seqüência e procura homologia no banco de dados. Descarta todas as pesquisas com pontuação pequena (score baixo) e vai alinhando a vizinhança das com pontuação boa, até chegar ao máximo valor. É fácil verificar que algumas regiões de certos genes alinham bem, mas outras pouco conservadas, não. O Alinhador Local não quer chegar ao alinhamento final, ele só quer identificar sequências com um nível de homologia significativo
 e vai alinhando a vizinhança das com pontuação boa, até chegar ao máximo valor. É fácil verificar que algumas regiões de certos genes alinham bem, mas outras pouco conservadas, não. O Alinhador Local não quer chegar ao alinhamento final, ele só quer identificar sequências com um nível de homologia significativo.")
21
Alinhamento local O fundamento teórico é que a função gênica está quase sempre confinada em domínios contínuos de uma proteína Se não fosse assim, não teria sentido usar...

22
Programas BLAST & Bancos
Há vários Programas BLAST úteis Alguns são usados quando a sua sequência é de nucleotídeos (BLASTn, BLASTx e tBLASTx) Outros são usados quando a sua seqüência é de aminoácidos (BLASTp) E vários bancos de dados para escolher (nr, pdb, dbEST, yeast, month, etc...) Ou usa-se limites [organism]
 Outros são usados quando a sua seqüência é de aminoácidos (BLASTp) E vários bancos de dados para escolher (nr, pdb, dbEST, yeast, month, etc...) Ou usa-se limites [organism]")
23
BLASTn e BLASTx A EST identifica o gene homólogo: BLASTn
A EST identifica proteína ortóloga de outro organismo - a evolução conservou a proteína enquanto o DNA divergiu: BLASTx BLASTx: a EST traduzida em seis proteínas 1 existe, 5 não... O mundo Blast é assim

24
tBLASTx tBLASTx traduz sua seqüência de nucleotídeos para proteína nas 6 possibilidades, exatamente como BLASTx Depois pesquisa com essas 6 proteínas deduzidas, um banco de dados de nucleotídeos também traduzido dessa maneira Pra que serve? Pois imagine que a telomerase de Euplotes seja parecida com a telomerase humana, mas os dois DNA não! Traduzindo a seqüência pesquisada e o banco de dados dbEST foi possível encontrar seqüências da telomerase humana

25
Aglomerados ou Clusters
Uma das atividades em bioinformática é formar aglomerados de todas as sequências geradas no projeto (as figurinhas de um álbum) Podemos saber quantas vezes um gene foi seqüenciado, e detectar os freqüentes! E quantos dos genes foram detectados Usa-se também para validar bibliotecas
 Podemos saber quantas vezes um gene foi seqüenciado, e detectar os freqüentes! E quantos dos genes foram detectados. Usa-se também para validar bibliotecas.")
26
Programas para aglomerar
Icatools Phrap Cap3, Cap4 Swat BLAST MegaBLAST Um aglomerado = Um gene

27
Qualidade das bibliotecas (100 primeiras ESTs)
Número de seqüências Boa biblioteca ? 1 2 3 4 5 7 9 11 Freqüência em que uma EST foi amostrada

28
UniGene Organização das sequências do GenBank em um conjunto de aglomerados Cada aglomerado do UniGene contém as sequências que representam um gene único E também informações relacionadas, como em que tecidos o gene é expresso, etc. E também onde está mapeado

29
MegaBLAST gera o UniGene
Todas ESTs contra todas Detecção de homologia > 96% de identidade > 70% do potencial Aglomerar

Apresentações semelhantes



 dos genes e seu armazenamento>")















>")