Carregar apresentação
A apresentação está carregando. Por favor, espere

1
Disciplina: Genética II
Profa. Dra. Ana Elizabete Silva Hemoglobinopatias

2
O que são as hemoglobinopatias ?
Distúrbios das hemoglobinas humanas Mutações nos genes das globinas Doença monogênica mais comum Anemia Hereditária

3
Qual o diagnóstico provável?
Caso Clínico 1 • Menino de raça negra, com 4 meses de idade; • Terceiro filho de pais jovens, naturais de Luanda; • O primeiro filho do casal faleceu aos 5 meses; • Aparentemente saudável até 15 dias antes do internamento, com febre e tosse; desde então ficou prostrado e progressivamente mais pálido. Foram realizados diversos exames complementares de diagnóstico, como hemograma com os seguintes resultados: Hemoglobina – 4,6g/dL (13 a 16g/dL) N.º Glóbulos vermelhos – /mm3 (4,5 a 6 milhões) N.º Glóbulos brancos – 8.000/uL (4.000 – ) N.º Plaquetas – /uL ( ) Qual o diagnóstico provável?
 N.º Glóbulos vermelhos – /mm3 (4,5 a 6 milhões) N.º Glóbulos brancos – 8.000/uL (4.000 – ) N.º Plaquetas – /uL ( ) Qual o diagnóstico provável")
4
Qual tipo de anemia? Ferropriva ou hereditária?
Solicitar esfregaço de sangue e eletroforese de hemoglobinas Teste de falcização positivo Esfregaço normal

5
Estrutura e função FUNÇÃO: transportador de oxigênio em hemácias de vertebrados. ESTRUTURA: tetrâmero 2 cadeias tipo alfa () 2 cadeias tipo beta () Cada subunidade : -cadeia polipeptídica globina grupo prostético (cofator): grupo Heme -
 Cada subunidade : -cadeia polipeptídica globina -grupo prostético (cofator): grupo Heme. -")
6
Formação da estrutura quaternária da hemoglobina.
4 cadeias são dobradas tetrâmero globular, com peso molecular de D

7
CARACTERÍSTICAS DA ESTRUTURA DAS GLOBINAS
- estrutura bem conservada ao longo da evolução Estrutura terciária: quase todas as globinas possuem as 8 regiões helicoidais A - H 2 aminoácidos histidina (resíduo F8) conservados fenilalanina (resíduo CD1) sítios internos: resíduos apolares invólucro hidrofóbico. superfície da molécula: resíduos polares (hidrofílicos) Mutações conformação/ substituição aa. conservados/alteração do invólucro hidrofóbico Hemoglobinopatias
 conservados fenilalanina (resíduo CD1) sítios internos: resíduos apolares invólucro hidrofóbico. superfície da molécula: resíduos polares (hidrofílicos) Mutações conformação/ substituição aa. conservados/alteração do invólucro hidrofóbico Hemoglobinopatias.")
8
CARACTERIZAÇÃO DAS CADEIAS DE GLOBINA
Cadeias e : tamanhos semelhantes cadeia 141aa cadeia 146 aa

9
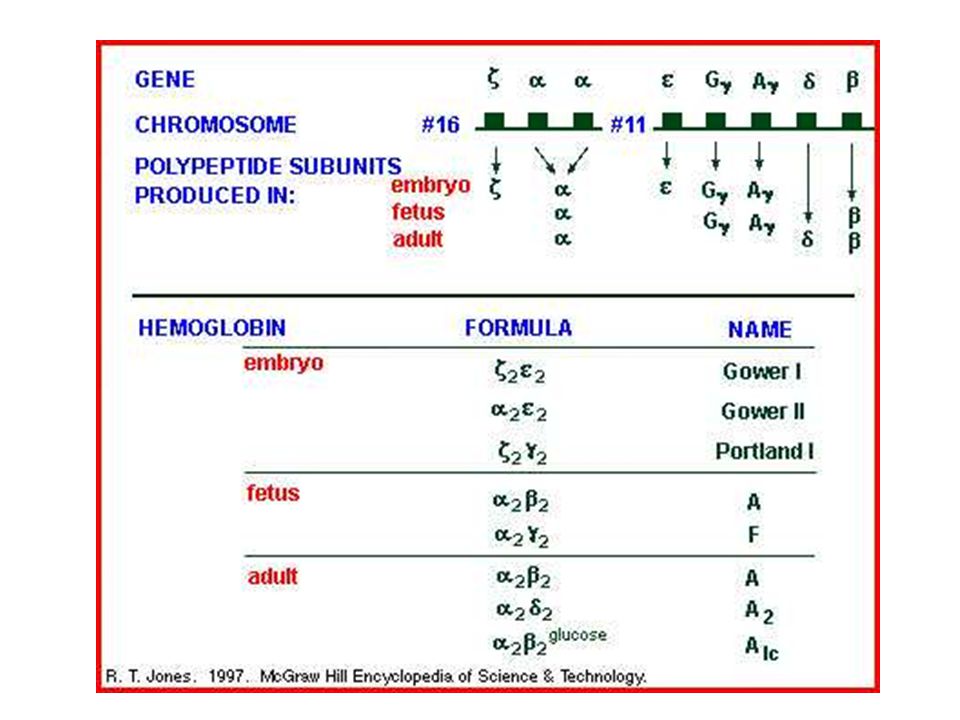
Genes das globinas, distribuídos em dois cromossomos.
genes da cadeia : cromossomo 16 ; 2; 1 - genes da cadeia : cromossomo 11 ; G; A; ; e diferem em 10 aa

10
Cluster Beta – LCR (REGIÃO CONTROLADORA DO LOCUS)
responsável pela expressão em alto nível do gene da -globina nos tecidos apropriados e durante o desenvolvimento: - controla expressão dos genes da família beta globina: atividade de estimulação, inibição interação de fatores de transcrição com o DNA e maquinaria da RNA polimerase Atua como um SUPERACENTUADOR de transcrição - LCR localizado ~20 kb do gene épsilon possui 5 sítios hipersensíveis (HS) -deleções da LCR não expressam os genes do grupo da -globina alvo para terapia gênica.
 -deleções da LCR não expressam os genes do grupo da -globina. alvo para terapia gênica.")
11
G A LCR Ativação dos Genes Beta Embrionário DNA Fetal DNA
Adulto DNA Strachan and Reald HMG2 1999

12
Como ocorre a ativação e silenciamento dos genes da globina durante o desenvolvimento?

13
ONTOGÊNIA – Tipos de globinas durante as fases do desenvolvimento

14
Hb Gower 1 Hb Gower 2 HbF HbA2 HbA

15
Expressão dos genes da globina durante o desenvolvimento
Mudanças temporais da síntese de globina alterações no local da eritropoiese. SÍNTESE DE GLOBINA EMBRIONÁRIA: - 3a à 8a semanas de gestação: eritropoiese no saco vitelínico Hb embrionárias globinas e (combinação com as demais) 3 Hb embrionárias Hb Gower 1 (2 2 ) Hb Gower 2 (2 2 ) Hb Portland (2 2 )
 3 Hb embrionárias Hb Gower 1 (2 2 ) Hb Gower 2 (2 2 ) Hb Portland (2 2 )")
16
Hb F SÍNTESE DA HEMOGLOBINA FETAL 5a semana gestação:
hematopoiese muda para o fígado fetal mudança na síntese de para e de para Hb fetal 2 2 (Hb F). -predominante (vida fetal) -70% da Hb total ao nascimento, mas na idade adulta 1%. Cadeias : - detectáveis no início da gestação síntese significativa próximo ao nascimento. Cadeia : -síntese inicia no final da vida fetal, continuando após nascimento, não atinge + de 2% da Hb adulta. Hb F
. -predominante (vida fetal) -70% da Hb total ao nascimento, mas na idade adulta 1%. Cadeias : - detectáveis no início da gestação síntese significativa próximo ao nascimento. Cadeia : -síntese inicia no final da vida fetal, continuando após nascimento, não atinge + de 2% da Hb adulta. Hb F.")
17
SÍNTESE DA HEMOGLOBINA ADULTA
-hematopoiese: medula óssea -Recém-nascido: HbF (2 2 ): 70-90% HbA (2 2): 0-10% -Após 6 meses de vida: hemoglobinas do adulto HbA (2 2): 95% HbA2 (2 2): 2-3% HbF (2 2 ): até 1%
: 70-90% HbA (2 2): 0-10% -Após 6 meses de vida: hemoglobinas do adulto. HbA (2 2): 95% HbA2 (2 2): 2-3% HbF (2 2 ): até 1%")
19
DOSAGEM GÊNICA E DOENÇAS CLÍNICAS
-globina : 4 genes globina : 2 genes -uma única mutação no gene -globina 25% das cadeias . -mutações da -globina doença severa na vida fetal e pós-natal. - uma única mutação no gene -globina afeta 50% das cadeias . - mutações da -globina não trazem consequências pré-natais (globina e a Hb F). diplóide
. diplóide.")
20
DISTÚRBIOS GENÉTICOS DA HEMOGLOBINA
A. Variantes estruturais: alteram a globina (mudanças na sequência de aminoácidos), sem afetar a taxa de síntese B. Talassemias: alteração na síntese desequilíbrio nas quantidades das cadeias do tipo alfa e beta Variantes estruturais: maioria provém de mutações de ponto. - > 900 variantes descritas - anemia hemolítica - transporte de O2 alterado - Brasil: mais comuns Hb S, Hb C e Hb D
, sem afetar a taxa de síntese. B. Talassemias: alteração na síntese desequilíbrio nas quantidades das cadeias do tipo alfa e beta. Variantes estruturais: maioria provém de mutações de ponto. - > 900 variantes descritas. - anemia hemolítica. - transporte de O2 alterado. - Brasil: mais comuns Hb S, Hb C e Hb D.")
21
HEMOGLOBINAS VARIANTES
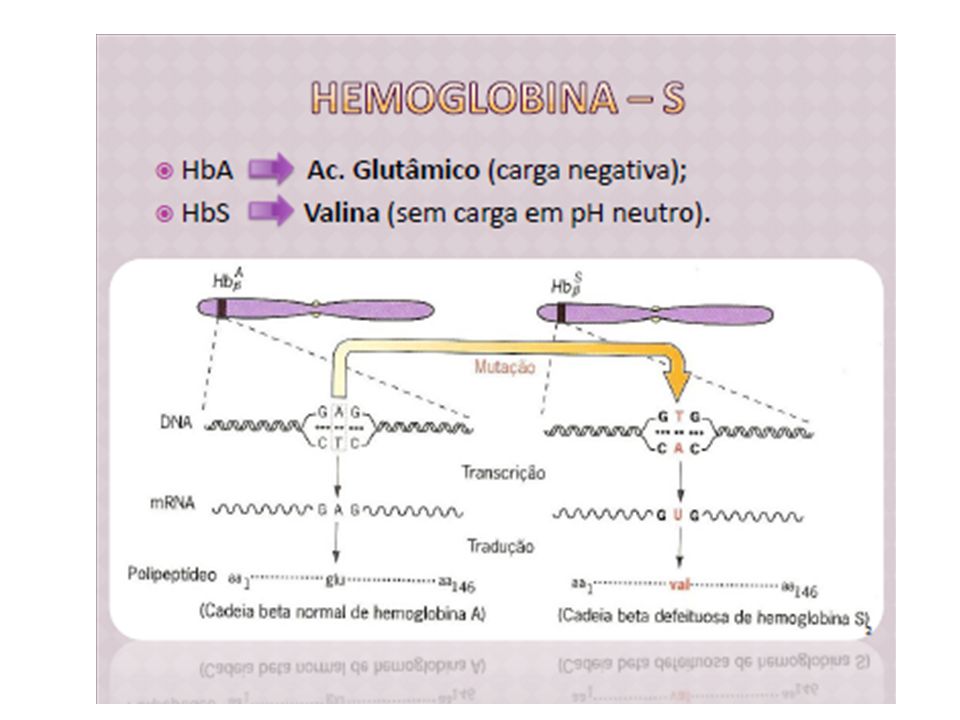
Hemoglobina S: Hb S (primeira Hb anormal detectada) alteração: 1 aa. dos 146 da -globina 6 Glu Val (A T) Mutação em homozigose Anemia falciforme.
 alteração: 1 aa. dos 146 da -globina. 6 Glu Val (A T) Mutação em homozigose Anemia falciforme.")
23
PATOLOGIA MOLECULAR DA HB S
Pauling et al. (1949): Hb A e Hb S por eletroforese Ingram (1956): substituição de 1 dos 146 aa. na cadeia da -globina. Ac. Glutâmico: carga negativa Valina: neutro → mobilidade eletroforética mais lenta da HbS
: Hb A e Hb S por eletroforese. Ingram (1956): substituição de 1 dos 146 aa. na cadeia da -globina. Ac. Glutâmico: carga negativa. Valina: neutro → mobilidade eletroforética mais lenta da HbS.")
24
Eletroforese ácida de hemoglobinas em gel de agarose
Eletroforese ácida de hemoglobinas em gel de agarose. (1) Hb ASF em sangue de recém-nascido; (2) Hb AS; (3) Hb AA. Devido ao processo físico-químico de eletroendosmose a Hb Fetal migra com maior rapidez que a Hb A. Eletroforese alcalina de hemoglobinas em gel de agarose. Diferenciação da mobilidade eletroforética dos genótipos SC, SF e AS.
 Hb ASF em sangue de recém-nascido; (2) Hb AS; (3) Hb AA. Devido ao processo físico-químico de eletroendosmose a Hb Fetal migra com maior rapidez que a Hb A. Eletroforese alcalina de hemoglobinas em gel de agarose. Diferenciação da mobilidade eletroforética dos genótipos SC, SF e AS.")
25
DISTRIBUIÇÃO GEOGRÁFICA
- África equatorial: 1/50 nascimentos - Região do Mediterrâneo - Índia e países onde essas pessoas migraram. - incidência: 1/400 a 1/600 afro-americanos. - hemácias: forma afoiçada ( tensão de O2)
")
26
AFOIÇAMENTO E SUAS CONSEQUÊNCIAS Hb com subunidades mutantes
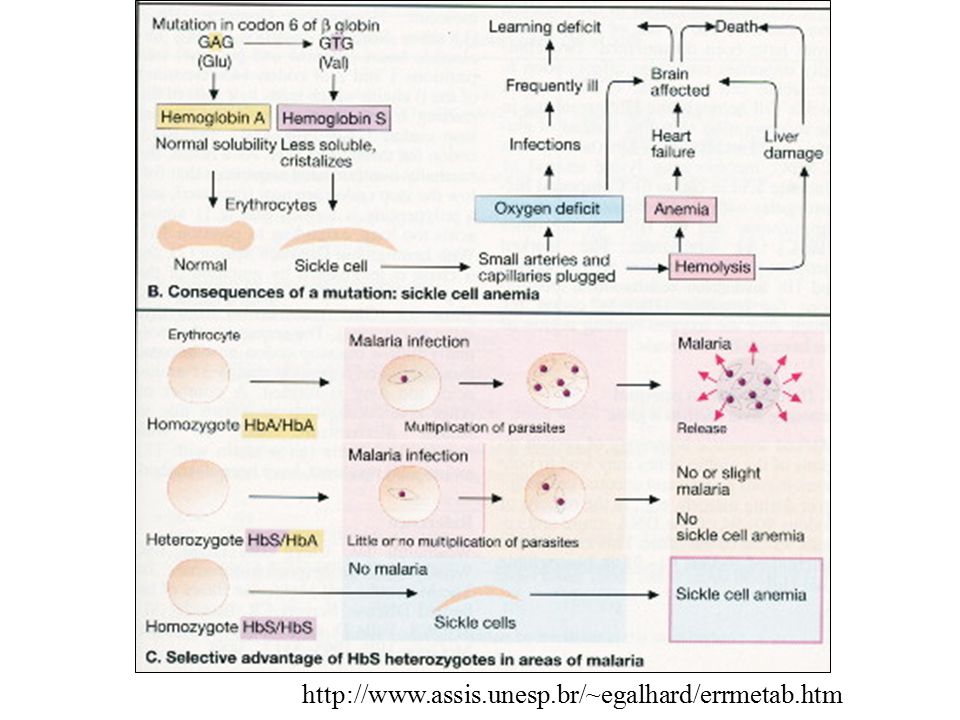
condições de tensão de O2 Hb S agregam-se em polímeros que distorcem o eritrócito (insolubilidade) - são menos flexíveis, não se comprimem nos capilares, obstruindo o fluxo sanguíneo hipóxia local.
 - são menos flexíveis, não se comprimem nos capilares, obstruindo o fluxo sanguíneo hipóxia local.")
27
Duas explicações possíveis para a seleção contra homozigotos mutantes:
Como explicar a alta frequência do alelo mutante (2S) em algumas regiões da África? Duas explicações possíveis para a seleção contra homozigotos mutantes: 1. Alta taxa mutação ou 2. Heterozigotos portadores mais adaptados certas situações ambientais: heterozigotos portadores (traço falcêmico - HbAS) maior vantagem seletiva que os homozigotos normais (HbAA).
 em algumas regiões da África Duas explicações possíveis para a seleção contra homozigotos mutantes: 1. Alta taxa mutação ou. 2. Heterozigotos portadores mais adaptados. certas situações ambientais: heterozigotos portadores (traço falcêmico - HbAS) maior vantagem seletiva que os homozigotos normais (HbAA).")
28
(HbAS) gene falcêmico : alta frequência em certas regiões da África heterozigotos são mais resistentes ao Plasmodium vivax (parte do ciclo evolutivo nas hemácias do hospedeiro)
")
31
MANIFESTAÇÕES CLÍNICAS
destruição prematura dos eritrócitos afoiçados anemia nos dois primeiros anos, atraso crescimento e atraso da puberdade, esplenomegalia, infecções repetidas (hipofunção esplênica), infartos nos tecidos (ulceração) problemas osteoarticulares e renais
, infartos nos tecidos (ulceração) problemas osteoarticulares e. renais.")
32
HEMOGLOBINA C - HbC substituição na 6a posição da cadeia (GAG AAG): 6 Glu Lys - menos solúvel que a Hb A cristaliza-se nas hemácias distúrbio hemolítico leve. - anemia sob estresse ou durante a gravidez gene C frequente na África ocidental e em descendentes dessa região. Hb SC: indivíduos possuem alelo S no outro locus da -globina. distúrbio hemolítico mais leve que a anemia falciforme

33
mutação de ponto substituição na cadeia 42 Phe Ser
HEMOGLOBINA INSTÁVEL HB HAMMERSMITH mutação de ponto substituição na cadeia 42 Phe Ser fenilalanina resíduo CD1 - aa conservados (alojar o Heme) -resíduo de serina é menor Heme sai do lugar - instabilidade ( hemoglobina precipita formando inclusões) - afinidade por O2 cianose.
 -resíduo de serina é menor Heme sai do lugar - instabilidade ( hemoglobina precipita formando inclusões) - afinidade por O2 cianose.")
34
HEMOGLOBINAS COM ALTERAÇÕES FUNCIONAIS TRANSPORTE DE O2 ALTERADO
-mutações que alteram a capacidade da Hb transportar O2 Metemoglobinas – Hb M Oxihemoglobina: forma normal da Hb em que o ferro está no estado reduzido (ferroso) Metemoglobina: o ferro encontra-se na forma férrica (oxidada) incapaz de transportar O2 cianose; assintomática nos heterozigotos. - Ex.: Hb Hyde Park - 92 His Tyr - histidina: aa conservado (resíduo F8)
 Metemoglobina: o ferro encontra-se na forma férrica (oxidada) incapaz de transportar O2 cianose; assintomática nos heterozigotos. - Ex.: Hb Hyde Park - 92 His Tyr. - histidina: aa conservado (resíduo F8)")
35
Hb Lepore - alguns pacientes com fenótipo de -talassemia:
- cadeias globínicas híbridas : metade N-terminal de uma cadeia fundida com a metade C-terminal de uma cadeia normal - Fusão da porção 5’ do gene com 3’ do gene pareamento desigual na MEIOSE crossing-over homólogo (genes homólogos)
")
36
Hb Lepore: cadeias fusionadas.
Pareamento normal Pareamento anormal Crossing-over desigual Anti-Lepore Lepore Hb Lepore: cadeias fusionadas.

37
- mutação provoca redução ou ausência na síntese de uma das cadeias ou , sem alterar a sequência de aminoácidos - cadeia produzida na taxa normal: excesso relativo precipitação na célula lesando a membrana destruição prematura da hemácia. TALASSEMIAS 2 grupos talassemias talassemias Distribuição geográfica ao redor do Velho mundo: Mediterrâneo, Oriente Médio e partes da África, Índia e Ásia. Thalassa: mar

38
Recombinação homóloga desigual
ALFA-TALASSEMIAS distúrbios genéticos na produção da -globina afetam a formação das Hb fetal (HbF 2 2 ) e adulta (HbA 2 2) Ausência de cadeia : talassemia 0 Redução de cadeia : talassemia + - ausência de cadeias de -globina: cadeias de -globina formam um tetrâmero. maioria das -talassemias deleções Recombinação homóloga desigual
 e adulta (HbA 2 2) Ausência de cadeia : talassemia 0. Redução de cadeia : talassemia + - ausência de cadeias de -globina: cadeias de -globina formam um tetrâmero. maioria das -talassemias deleções. Recombinação homóloga desigual.")
39
Formas de Talassemia Alfa
Deleção 4 genes alfa: formação de 4 Hb Barts forma major hidropisia fetal Deleção de 3 genes alfa: tetrâmero 4 Doença da Hb H precipitam na hemácia destruição e anemia hemolítica Deleção de 2 genes alfa: anemia leve (traço talassêmico) Deleção de 1 gene alfa: portador silencioso
 Deleção de 1 gene alfa: portador silencioso.")
40
- estado heterozigoto (2 genes normais e 2 mutantes).
- -talassemia por deleção homozigota (ausência de cadeias ) restrita ao SE asiático. - estado heterozigoto (2 genes normais e 2 mutantes). 2 genótipos —/ — — —/ progênie — —/— — Hidropisia fetal Pais Hidropisia Fetal
 restrita ao SE asiático. - estado heterozigoto (2 genes normais e 2 mutantes). 2 genótipos —/ — — —/ progênie — —/— — Hidropisia fetal. Pais. Hidropisia Fetal.")
41
DOENÇA DA HB H Precipitados da HbH
Doença da Hb H (4): deleção de 3 genes (_ _/ _ ) moderadamente grave. alteracões ósseas, esplenomegalia afeta orientais e brancos Pais Hemoglobina H Traço -talassêmico: deleção de 2 genes (_ _/ ) : - anemia leve. - comum em orientais e SE asiático Precipitados da HbH
: deleção de 3 genes (_ _/ _ ) moderadamente grave. alteracões ósseas, esplenomegalia. afeta orientais e brancos. Pais. Hemoglobina H. Traço -talassêmico: deleção de 2 genes (_ _/ ) : - anemia leve. - comum em orientais e SE asiático. Precipitados da HbH.")
42
SO Ásia, negros americanos e brasileiros
TRAÇO ALFA-TALASSÊMICO Traço -talassêmico (_/_ ) SO Ásia, negros americanos e brasileiros Pais descendentes
 SO Ásia, negros americanos e brasileiros. Pais. descendentes.")
43
DESEQUILÍBRIOS DAS GOBINAS ALFA
(4) (4)
 (4)")
44
Ausência de cadeia : talassemia 0
BETA-TALASSEMIAS produção reduzida ou ausente de -globina anemia microcítica hipocrômica. Ausência de cadeia : talassemia 0 Redução de cadeia : talassemia + desequilíbrio da síntese de -globinas precipitação das cadeias danos na hemácia. - cadeia é importante no período pós-natal síntese após o nascimento (-globina substituiria a e a síntese de Hb A é ). - maioria das -talassemias são decorrentes de mutações pontuais síntese de RNA - síntese de Hb F reduz a gravidade da doença. indivíduos homozigotos para -talassemia talassemia major indivíduos heterozigotos para -talassemia talassemia minor
. - maioria das -talassemias são decorrentes de mutações pontuais síntese de RNA. - síntese de Hb F reduz a gravidade da doença. indivíduos homozigotos para -talassemia talassemia major. indivíduos heterozigotos para -talassemia talassemia minor.")
45
1. Mutação nos promotores (TATA box)
2. Mutação no splicing do mRNA 3. Mutação da cobertura (cap) do mRNA 4. Mutações sem sentido e de matriz de leitura
 do mRNA. 4. Mutações sem sentido e de matriz de leitura.")
46
Normal Hipocrômica e microcítica

47
DESEQUILÍBRIOS DAS GOBINAS BETA
Figura 7.13: Relação entre quantidade de globinas alfa livre, níveis de Hb A, Hb A2 e Hb Fetal, e gravidade do quadro clínico em pacientes com talassemia beta menor e maior. Figura 7.13: Relação entre quantidade de globinas alfa livre, níveis de Hb A, Hb A2 e Hb Fetal, e gravidade do quadro clínico em pacientes com talassemia beta menor e maior. DESEQUILÍBRIOS DAS GOBINAS BETA

48
TIPOS DE TALASSEMIA BETA

49
-anemia intensa antes dos 2 anos (Hb F pós-natal),
TALASSEMIA MAJOR – ANEMIA DE COOLEY Sinais Clínicos -anemia intensa antes dos 2 anos (Hb F pós-natal), -atraso do crescimento, -icterícia, -hepatoesplenomegalia, -alterações esqueléticas (expansão da medula óssea). Tratamento - transfusão sanguínea - administração de agentes quelantes do ferro transplante de medula óssea Terapia gênica
, -atraso do crescimento, -icterícia, -hepatoesplenomegalia, -alterações esqueléticas (expansão da medula óssea). Tratamento. - transfusão sanguínea. - administração de agentes quelantes do ferro. transplante de medula óssea. Terapia gênica.")
51
TALASSEMIA MAJOR – ANEMIA DE COOLEY
Eletroforese de hemoglobinas em gel de agarose alcalina: (1) Hb AF de talassemia beta maior; (2) Hb S/beta talassemia ou SF, onde m indica o fracionamento da metaemoglobina S e a a globina alfa livre; (3) Hb AA2 e Fetal em tal. beta menor; (4) Hb AS (traço falciforme); (5) Hb AA (normal).
 Hb AF de talassemia beta maior; (2) Hb S/beta talassemia ou SF, onde m indica o fracionamento da metaemoglobina S e a a globina alfa livre; (3) Hb AA2 e Fetal em tal. beta menor; (4) Hb AS (traço falciforme); (5) Hb AA (normal).")
52
Cadeias normais em excesso precipitam-se formando os corpúsculos de Heinz na hemácia em forma de lágrima.

53
TALASSEMIA MINOR -TRAÇO TALASSÊMICO
portadores de um alelo mutante (heterozigoto) Geralmente assintomáticos - anemia leve diagnóstico: eletroforese ( Hb A2).
 Geralmente assintomáticos. - anemia leve. diagnóstico: eletroforese. ( Hb A2).")
54
Risco para casamentos entre portadores de talassemia beta

55
DIAGNÓSTICO LABORATORIAL
Eletroforese em acetato de celulose (pH alcalino ou ácido): rápida e baixo custo, mas sensibilidade limitada Hbs co-migram na mesma posição.
: rápida e baixo custo, mas sensibilidade limitada Hbs co-migram na mesma posição.")
56
DIAGNÓSTICO LABORATORIAL
DIAGNÓSTICO LABORATORIAL Eletroforese de Focalização isoelétrica: técnica de alta precisão identifica Hbs com co-migração A Hb migra no gel e se precipita ao encontrar seu ponto isoelétrico (conforme o pH no gradiente do gel)
")
57
DIAGNÓSTICO LABORATORIAL
HPLC (Cromatografia líquida de alta pressão): diagnóstico diferencial das hemoglobinopatias (dados quantitativos e qualitativos) Técnicas moleculares: identificação da mutação (PCR, sequenciamento)
: diagnóstico diferencial das hemoglobinopatias (dados quantitativos e qualitativos) Técnicas moleculares: identificação da mutação (PCR, sequenciamento)")
58
5- Considerando que a amostra número 1 é o perfil adulto normal (HbAA) responda quais são os perfis das amostras 2 e 3 nas eletroforeses A e B. Eletroforese A Eletroforese B - + A F S e similares A2; C e similares - + A F S e similares A2; C e similares

Apresentações semelhantes